Clear Sky Science · de
End-to-end Deep-Attention-Multitask-Pipeline zur Vorhersage von Unsicherheits-quantifizierten Peptid-Eigenschaften aus Massenspektrometrie-Daten
Warum diese Forschung für Gesundheit und Biologie wichtig ist
Moderne biomedizinische Forschung stützt sich stark auf Massenspektrometrie, um zu ermitteln, welche Proteine in Zellen und Geweben vorhanden sind. Dennoch bleibt trotz leistungsfähiger Instrumente und großer Datenbanken ein überraschender Teil der Messungen ungeklärt, insbesondere bei seltenen oder ungewöhnlichen Proteinen, die für Krankheiten wie Krebs oder neurologische Störungen von Bedeutung sein können. Diese Arbeit stellt ProteoRift vor, ein maschinelles Lernsystem, das hilft, mehr von diesen verborgenen Informationen zu entdecken, indem es zentrale Eigenschaften von Proteinfragmenten direkt aus Rohdaten vorhersagt und zugleich Wissenschaftlern angibt, wie zuversichtlich diese Vorhersagen sind.

Der Engpass beim Auslesen von Protein-Fingerprints
Massenspektrometrie funktioniert, indem Proteine in kleinere Stücke, sogenannte Peptide, zerlegt werden und die Masse der resultierenden Fragmente gemessen wird. Standardsoftware durchsucht dann große Proteindatenbanken nach Peptidsequenzen, deren berechnete Masse zu jedem beobachteten Spektrum passt. Um diese Suche rechnerisch handhabbar zu machen, wenden die meisten Werkzeuge eine einfache Regel an: Sie betrachten nur Kandidaten, deren Gesamtmasse eng mit dem gemessenen Wert übereinstimmt. Diese massenbasierte Filterung beschleunigt den Prozess, hat aber einen Preis. Wird die Masse geringfügig falsch zugewiesen oder trägt ein Peptid eine unerwartete chemische Modifikation, kann die richtige Antwort bereits ausgeschlossen werden, bevor sie überhaupt in Betracht gezogen wird, was zum großen Bestand nicht zugeordneter Spektren und zu einer Verzerrung zugunsten häufiger, gutartiger Peptide beiträgt.
Eine klügere Methode zur Einschränkung der Suche
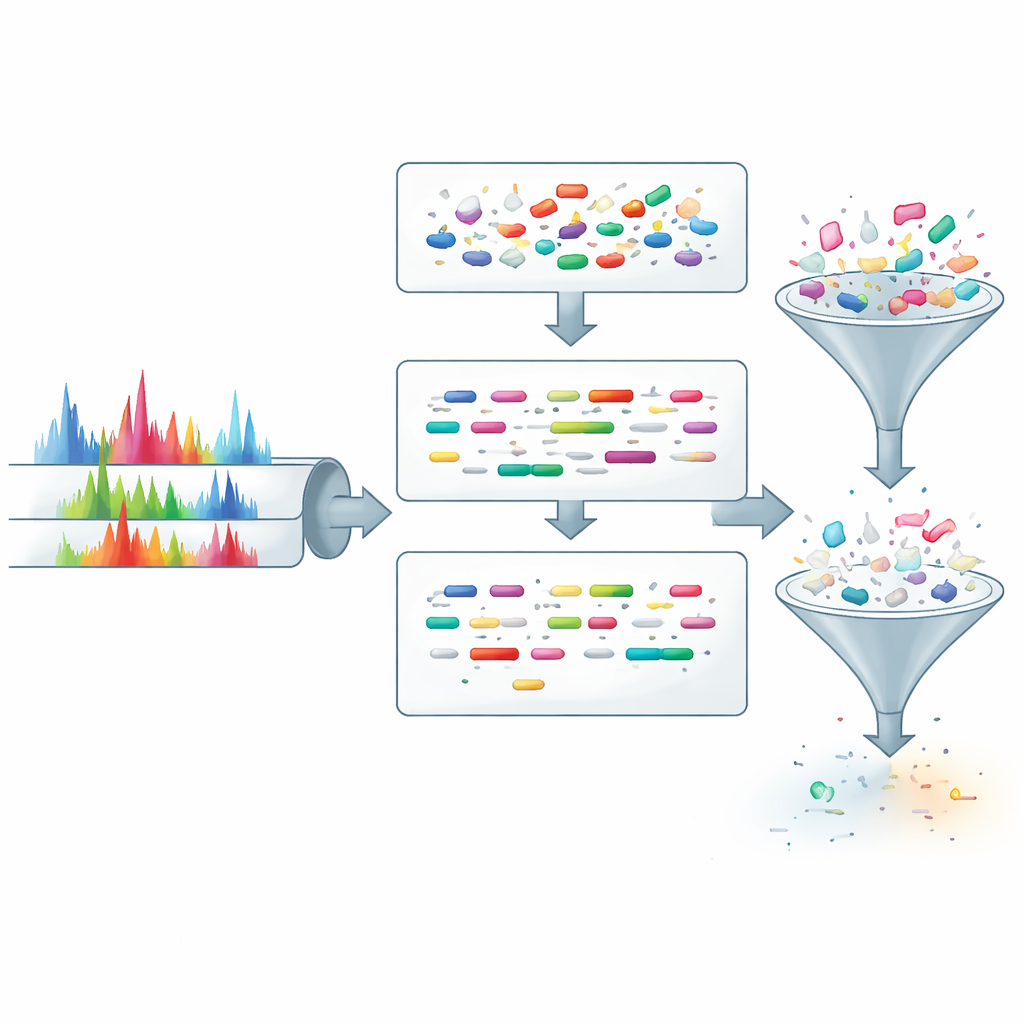
ProteoRift schlägt eine andere Strategie vor: Anstatt Kandidaten nur nach Masse zu filtern, lernt das System, aus jedem Spektrum reichhaltigere Informationen zu extrahieren, bevor eine Datenbanksuche stattfindet. Das System basiert auf einem auf Attention beruhenden tiefen neuronalen Netzwerk, das als Eingabe das Muster der Peaks in einem Spektrum zusammen mit grundlegenden Aufnahmeparametern erhält. Daraus sagt es gleichzeitig drei Eigenschaften des zugrundeliegenden Peptids voraus: seine Länge, wie oft es bei der Probenvorbereitung geschnitten wurde (missed cleavages) und ob es Modifikationen trägt. Da diese Aufgaben zusammenhängen, fördert ihre gemeinsame Ausbildung, dass das Modell eine robuste interne Darstellung von Spektren bildet, was seine Fähigkeit verbessert, auf neue Daten zu generalisieren.
Vorhersagen in schnellere und schlankere Suchen verwandeln
Um diese Vorhersagen nutzbar zu machen, integrieren die Autoren ProteoRift in eine End-to-End-Pipeline zusammen mit einem zuvor entwickelten Werkzeug namens SpeCollate, das Spektren mit Peptidsequenzen in einem Einbettungsraum abgleicht. Zuerst weist ProteoRift jedes Spektrum einer Klasse zu, definiert durch Längenbereich, Anzahl der verpassten Schnitte und Modifikationsstatus. Peptide in der Datenbank werden ähnlich anhand ihrer bekannten Eigenschaften gruppiert. Die Suchmaschine vergleicht dann Spektren nur mit Peptiden derselben Klasse, anstatt jedes Peptid mit ähnlicher Masse zu durchlaufen. In mehreren menschlichen und Mikrobiom-Datensätzen verkleinert diese gezielte Filterung den Kandidatenraum theoretisch um mehr als 90 % und liefert praktische Beschleunigungen von etwa dem 8- bis 12-Fachen gegenüber nur massenbasierten Filtern, während eine ähnliche Anzahl sicher identifizierter Peptide erhalten bleibt. In sehr großen proteogenomischen und metaproteomischen Datenbanken können die Beschleunigungen in bestimmten Tests sogar noch höher sein und über das 40-Fache erreichen.
Wissen, wann das Modell falsch liegen könnte
Da Systeme des maschinellen Lernens häufig als Blackboxen betrachtet werden, entwickeln die Autoren zudem Unsicherheitsmaße, die speziell auf Massenspektrometrie-Daten zugeschnitten sind. Sie untersuchen, wie stark sich die interne Darstellung eines Spektrums unter kontrollierten Verzerrungen verändert, wie dicht es von ähnlichen Trainingsbeispielen umgeben ist und wie gut die Struktur der Originaldaten im gelernten Raum erhalten bleibt. Diese drei Metriken erfassen unterschiedliche Aspekte von Unsicherheit: Messrauschen und Lücken in dem, was das Modell während des Trainings gesehen hat. Kombiniert können sie vertraute von unbekannten Daten mit sehr hoher Genauigkeit unterscheiden und helfen, Fälle zu kennzeichnen, in denen die bestbewertete Peptid-Übereinstimmung des Modells wahrscheinlich korrekt ist.
Was das für zukünftige Entdeckungen bedeutet
Alltagssprachlich funktioniert ProteoRift wie ein intelligenter Türwächter, der ein Spektrum betrachtet und sagt: „Das ist wahrscheinlich ein kurzes, unverändertes Peptid mit einem Schnitt,“ oder „das sieht länger und modifiziert aus,“ und dann nur passende Kandidaten in die detaillierte Suche lässt. Dadurch beschleunigt es die Analyse deutlich, ohne viel Genauigkeit einzubüßen, selbst bei komplexen oder sehr großen Proteindatenbanken. Gleichzeitig geben seine Unsicherheitsmetriken Forschern ein klareres Gefühl dafür, wann ein Ergebnis vertrauenswürdig ist oder wann mehr Daten oder eine Modellanpassung nötig sein könnten. Zusammen könnten diese Fortschritte dazu beitragen, die Massenspektrometrie über ihren derzeitigen Schwerpunkt auf häufige, gut charakterisierte Proteine hinaus zu bringen und neue Einblicke in seltene und modifizierte Peptide zu eröffnen, die oft die interessantesten biologischen Hinweise liefern.
Zitation: Tariq, U., Shabbir, B. & Saeed, F. End-to-end deep attention-based multitask pipeline for predicting uncertainty-quantified peptide properties from mass spectrometry data. Sci Rep 16, 13331 (2026). https://doi.org/10.1038/s41598-026-43215-2
Schlüsselwörter: Proteomik, Massenspektrometrie, Tiefes Lernen, Peptid-Identifikation, Unsicherheitsschätzung