Clear Sky Science · fr
Pipeline multitâche de bout en bout à base d’attention profonde pour prédire des propriétés de peptides quantifiées en incertitude à partir de données de spectrométrie de masse
Pourquoi cette recherche est importante pour la santé et la biologie
La recherche biomédicale moderne repose largement sur la spectrométrie de masse pour déterminer quels protéines sont présentes dans nos cellules et tissus. Pourtant, malgré des instruments puissants et de larges bases de données, une fraction surprenante des données reste inexpliquée, en particulier pour les protéines rares ou inhabituelles qui peuvent être cruciales dans des maladies comme le cancer ou les affections neurologiques. Cet article présente ProteoRift, un système d’apprentissage automatique qui aide à révéler davantage de ces informations cachées en prédisant des propriétés clés des fragments protéiques directement à partir des données brutes, tout en indiquant aux scientifiques le degré de confiance associé à ces prédictions.

Le goulot d’étranglement dans la lecture des empreintes protéiques
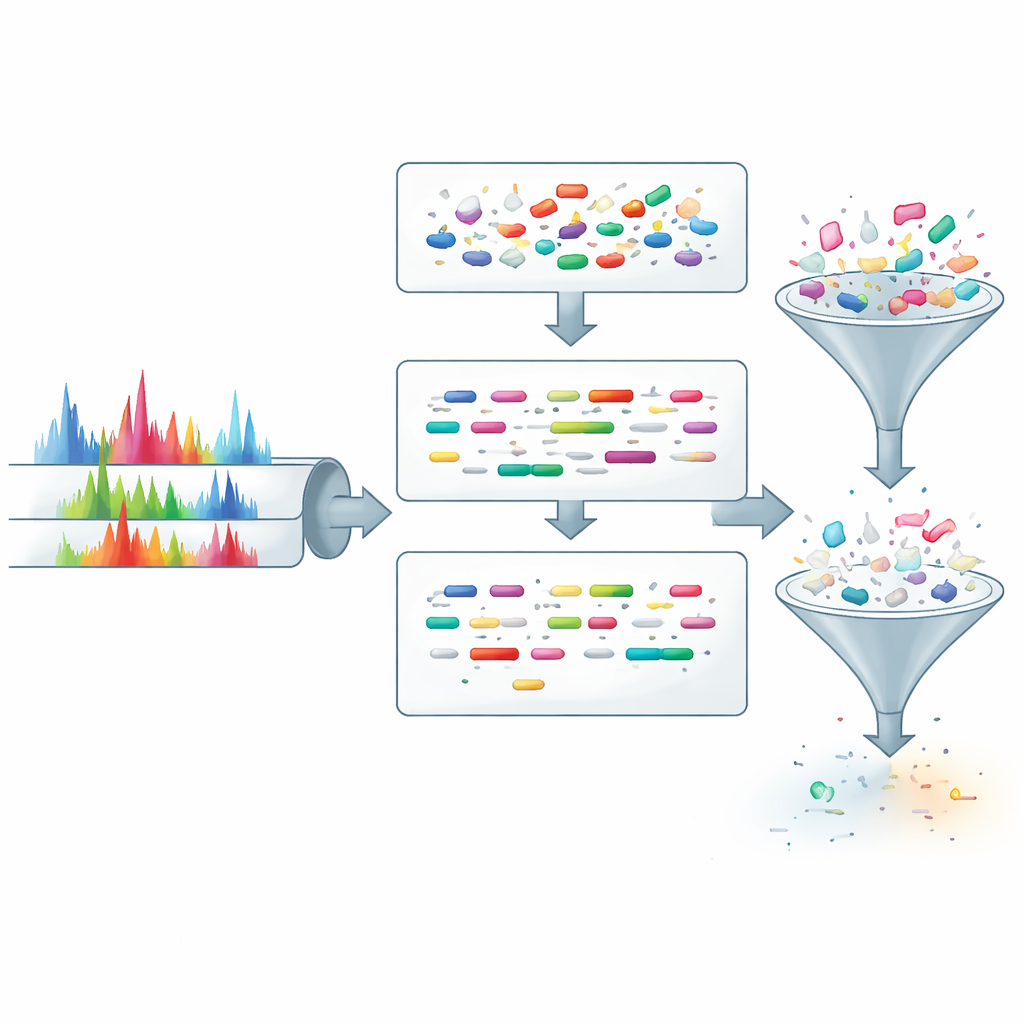
La spectrométrie de masse fonctionne en coupant les protéines en fragments plus petits appelés peptides et en mesurant la masse des fragments résultants. Les logiciels standards recherchent ensuite dans de larges bases de données de protéines des séquences peptidiques dont la masse calculée correspond à chaque spectre observé. Pour rendre cette recherche computable, la plupart des outils appliquent une règle simple : ils ne considèrent que les candidats dont la masse globale correspond étroitement à la valeur mesurée. Ce filtrage basé sur la masse accélère le processus, mais au prix d’un inconvénient. Si la masse est légèrement mal attribuée, ou si un peptide porte une modification chimique inattendue, la bonne réponse peut être exclue avant même d’être envisagée, contribuant au grand nombre de spectres non attribués et à un biais en faveur des peptides abondants et bien comportés.
Une façon plus intelligente de restreindre la recherche
ProteoRift propose une stratégie différente : au lieu de filtrer les candidats uniquement par la masse, il apprend à extraire des informations plus riches de chaque spectre avant toute recherche en base. Le système s’appuie sur un réseau neuronal profond à mécanisme d’attention qui prend en entrée le profil des pics d’un spectre ainsi que des détails d’acquisition basiques. À partir de cela, il prédit simultanément trois propriétés du peptide sous-jacent : sa longueur, le nombre de coupures manquées lors de la préparation de l’échantillon, et la présence éventuelle de modifications. Parce que ces tâches sont corrélées, les entraîner conjointement pousse le modèle à former une représentation interne robuste des spectres, améliorant sa capacité à généraliser à de nouvelles données.
Transformer les prédictions en recherches plus rapides et plus économes
Pour exploiter ces prédictions, les auteurs intègrent ProteoRift dans une chaîne de traitement de bout en bout aux côtés d’un outil développé précédemment appelé SpeCollate, qui associe spectres et séquences peptidiques dans un espace d’embeddings. D’abord, ProteoRift assigne chaque spectre à une classe définie par une plage de longueurs, le nombre de coupures manquées et le statut de modification. Les peptides de la base de données sont groupés de la même manière selon leurs propriétés connues. Le moteur de recherche compare alors uniquement les spectres aux peptides de la même classe, au lieu de balayer tous les peptides de masse similaire. Sur plusieurs jeux de données humains et microbiomes, ce filtrage ciblé réduit en théorie l’espace de recherche de candidats de plus de 90 % et offre des accélérations pratiques d’environ 8 à 12 fois par rapport aux filtres basés uniquement sur la masse, tout en retrouvant des nombres comparables de peptides identifiés avec confiance. Dans certaines bases de données protéogénomiques et méta-protéomiques très volumineuses, les gains de vitesse peuvent être encore supérieurs, dépassant 40 fois dans des tests spécifiques.
Savoir quand le modèle peut se tromper
Parce que les systèmes d’apprentissage automatique sont souvent perçus comme des boîtes noires, les auteurs développent aussi des mesures d’incertitude adaptées aux données de spectrométrie de masse. Ils sondent dans quelle mesure la représentation interne d’un spectre change sous des distorsions contrôlées, à quelle densité il est entouré d’exemples d’entraînement similaires, et dans quelle mesure la structure des données d’origine est préservée dans l’espace appris. Ces trois métriques capturent différents aspects de l’incertitude : le bruit dans les mesures elles-mêmes et les lacunes dans ce que le modèle a vu lors de l’entraînement. Combinées, elles peuvent distinguer données familières et non familières avec une très grande précision et aider à signaler les cas où l’accord de peptide le mieux noté par le modèle a de fortes chances d’être correct.
Ce que cela signifie pour les découvertes futures
De manière concrète, ProteoRift fonctionne comme un gardien intelligent qui regarde un spectre et dit « ceci est probablement un peptide court, non modifié, avec une coupure », ou « ceci semble plus long et modifié », puis n’autorise que les candidats appropriés dans la recherche détaillée. Ce faisant, il accélère considérablement l’analyse sans sacrifier beaucoup de précision, même sur des bases de données protéiques complexes ou très grandes. Parallèlement, ses métriques d’incertitude offrent aux chercheurs une meilleure appréciation du moment où il faut faire confiance à un résultat ou quand davantage de données ou un ajustement du modèle peuvent être nécessaires. Ensemble, ces avancées pourraient aider la spectrométrie de masse à dépasser son focus actuel sur les protéines abondantes et bien caractérisées et ouvrir de nouvelles fenêtres sur les peptides rares et modifiés qui recèlent souvent les indices biologiques les plus intéressants.
Citation: Tariq, U., Shabbir, B. & Saeed, F. End-to-end deep attention-based multitask pipeline for predicting uncertainty-quantified peptide properties from mass spectrometry data. Sci Rep 16, 13331 (2026). https://doi.org/10.1038/s41598-026-43215-2
Mots-clés: protéomique, spectrométrie de masse, apprentissage profond, identification de peptides, estimation de l’incertitude