Clear Sky Science · pl
End-to-end głęboki wielozadaniowy system oparty na mechanizmie uwagi do przewidywania właściwości peptydów z kwantyfikacją niepewności na podstawie danych z spektrometrii mas
Dlaczego te badania są ważne dla zdrowia i biologii
Współczesne badania biomedyczne w dużej mierze opierają się na spektrometrii mas do odczytywania, które białka są obecne w naszych komórkach i tkankach. Mimo zaawansowanych urządzeń i rozległych baz danych zaskakujący odsetek danych pozostaje jednak niewyjaśniony, zwłaszcza w przypadku rzadkich lub nietypowych białek, które mogą mieć kluczowe znaczenie w chorobach takich jak nowotwory czy zaburzenia neurologiczne. W artykule tym przedstawiono ProteoRift — system uczenia maszynowego, który pomaga odsłonić większą część tych ukrytych informacji, przewidując istotne właściwości fragmentów białek bezpośrednio z surowych danych, jednocześnie informując naukowców o pewności tych przewidywań.

Wąskie gardło w odczytywaniu odcisków białkowych
Spektrometria mas działa przez rozbijanie białek na mniejsze fragmenty zwane peptydami i mierzenie masy powstałych fragmentów. Standardowe oprogramowanie następnie przeszukuje duże bazy białek w poszukiwaniu sekwencji peptydów, których obliczona masa odpowiada obserwowanemu widmu. Aby zachować wykonalność obliczeniową, większość narzędzi stosuje prostą regułę: rozważa tylko kandydatów, których masa ogólna ściśle pasuje do wartości zmierzonej. Taka filtracja oparta na masie przyspiesza działanie, ale ma swoją cenę. Jeśli masa zostanie nieznacznie błędnie przypisana lub jeśli peptyd nosi nieoczekiwaną modyfikację chemiczną, poprawna odpowiedź może zostać wykluczona zanim w ogóle zostanie rozważona, co przyczynia się do dużej puli nieprzypisanych widm i do uprzedzenia wobec obfitych, „dobrze zachowujących się” peptydów.
Sprytniejszy sposób zawężania poszukiwań
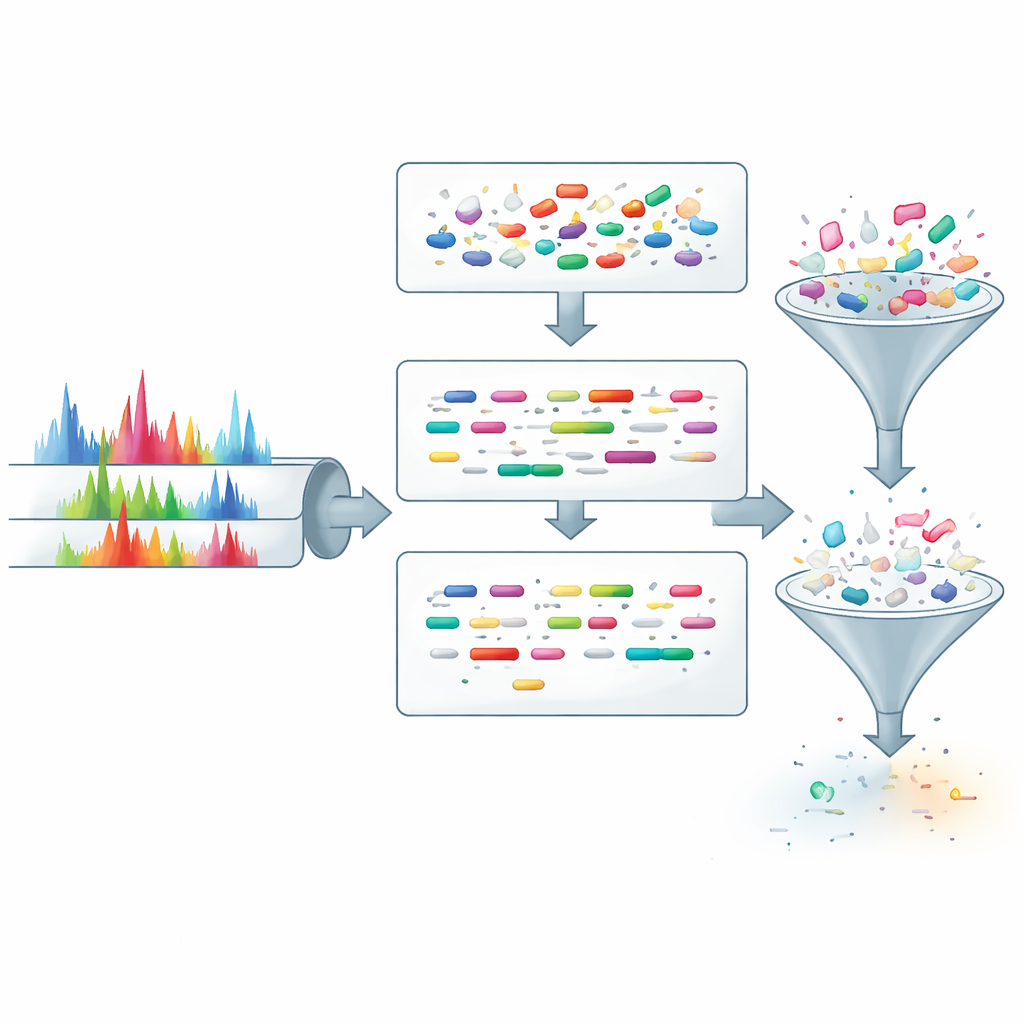
ProteoRift proponuje inną strategię: zamiast filtrować kandydatów wyłącznie na podstawie masy, uczy się wydobywać bogatsze informacje z każdego widma zanim nastąpi jakiekolwiek przeszukiwanie bazy. System opiera się na głębokiej sieci neuronowej z mechanizmem uwagi, która jako wejście przyjmuje wzorzec pików w widmie wraz z podstawowymi danymi akwizycji. Z tego jednocześnie przewiduje trzy właściwości leżącego u podstaw peptydu: jego długość, liczbę miejsc, w których nie nastąpiło przecięcie podczas przygotowania próbki (pominięte cięcia) oraz obecność modyfikacji. Ponieważ te zadania są ze sobą powiązane, trenowanie ich razem zachęca model do tworzenia odpornej wewnętrznej reprezentacji widm, poprawiając jego zdolność do uogólniania na nowe dane.
Przekuwanie przewidywań w szybsze i oszczędniejsze wyszukiwania
Aby wykorzystać te przewidywania w praktyce, autorzy integrują ProteoRift w całościowym pipeline razem z wcześniej opracowanym narzędziem SpeCollate, które dopasowuje widma do sekwencji peptydów w przestrzeni osadzeń. Najpierw ProteoRift przypisuje każde widmo do klasy zdefiniowanej zakresem długości, liczbą pominiętych przecięć i statusem modyfikacji. Peptydy w bazie są podobnie grupowane na podstawie znanych właściwości. Silnik wyszukiwania porównuje wtedy widma tylko z peptydami z tej samej klasy, zamiast przeszukiwać każdy peptyd o podobnej masie. W wielu zestawach danych ludzkich i mikrobiomu to ukierunkowane filtrowanie teoretycznie zmniejsza przestrzeń kandydatów o ponad 90% i w praktyce przyspiesza wyszukiwanie około 8- do 12-krotnie w porównaniu z filtrami opartymi wyłącznie na masie, przy jednoczesnym odzyskaniu podobnych liczby pewnie zidentyfikowanych peptydów. W niektórych bardzo dużych bazach proteogenomicznych i metaproteomicznych przyspieszenia mogą być jeszcze wyższe, osiągając w konkretnych testach ponad 40-krotność.
Wiedza, kiedy model może się mylić
Ponieważ systemy uczenia maszynowego są często postrzegane jako czarne skrzynki, autorzy opracowali również miary niepewności dostosowane do danych ze spektrometrii mas. Badają, jak bardzo wewnętrzna reprezentacja widma zmienia się pod kontrolowanymi zniekształceniami, jak gęsto jest otoczone podobnymi przykładami z zestawu treningowego oraz jak dobrze struktura oryginalnych danych jest zachowana w wyuczonej przestrzeni. Te trzy metryki wychwytują różne aspekty niepewności: szum w samych pomiarach oraz luki w tym, co model widział podczas treningu. W połączeniu potrafią bardzo dokładnie odróżnić dane znane od nieznanych i pomagają wskazywać przypadki, w których dopasowanie peptydu z najwyższym wynikiem przez model prawdopodobnie jest poprawne.
Co to oznacza dla przyszłych odkryć
Mówiąc prosto, ProteoRift działa jak inteligentny strażnik, który patrzy na widmo i mówi: „to prawdopodobnie krótki, niemodyfikowany peptyd z jednym cięciem” albo „to wygląda na dłuższy i zmodyfikowany”, i wpuści do szczegółowego wyszukiwania tylko odpowiednich kandydatów. Dzięki temu znacznie przyspiesza analizę, nie poświęcając przy tym wiele z dokładności, nawet na skomplikowanych lub bardzo dużych bazach białek. Równocześnie jego metryki niepewności dają badaczom jaśniejszy obraz, kiedy można ufać wynikowi, a kiedy potrzebne są dodatkowe dane lub dopracowanie modelu. Razem te postępy mogą pomóc przenieść spektrometrię mas poza obecne skupienie na obfitych, dobrze scharakteryzowanych białkach i otworzyć nowe okna na rzadkie i modyfikowane peptydy, które często zawierają najbardziej interesujące biologiczne wskazówki.
Cytowanie: Tariq, U., Shabbir, B. & Saeed, F. End-to-end deep attention-based multitask pipeline for predicting uncertainty-quantified peptide properties from mass spectrometry data. Sci Rep 16, 13331 (2026). https://doi.org/10.1038/s41598-026-43215-2
Słowa kluczowe: proteomika, spektrometria mas, głębokie uczenie, identyfikacja peptydów, szacowanie niepewności