Clear Sky Science · es
Tubería multitarea de atención profunda de extremo a extremo para predecir propiedades peptídicas cuantificadas con incertidumbre a partir de datos de espectrometría de masas
Por qué esta investigación importa para la salud y la biología
La investigación biomédica moderna depende en gran medida de la espectrometría de masas para determinar qué proteínas están presentes en nuestras células y tejidos. Sin embargo, a pesar de instrumentos potentes y grandes bases de datos, una fracción sorprendente de los datos permanece sin explicar, especialmente en el caso de proteínas raras o inusuales que pueden ser clave en enfermedades como el cáncer o trastornos neurológicos. Este artículo presenta ProteoRift, un sistema de aprendizaje automático que ayuda a descubrir más de esa información oculta al predecir propiedades clave de fragmentos proteicos directamente a partir de datos en bruto, al tiempo que informa a los científicos sobre la confianza de esas predicciones.

El cuello de botella en la lectura de huellas proteicas
La espectrometría de masas funciona rompiendo las proteínas en piezas más pequeñas llamadas péptidos y midiendo la masa de los fragmentos resultantes. El software estándar busca luego en grandes bases de datos de proteínas secuencias de péptidos cuya masa calculada coincida con cada espectro observado. Para que esta búsqueda sea computacionalmente factible, la mayoría de las herramientas aplican una regla simple: solo consideran candidatos cuya masa total coincide estrechamente con el valor medido. Este filtrado basado en la masa acelera el proceso, pero tiene un coste. Si la masa se asigna ligeramente mal, o si un péptido porta una modificación química inesperada, la respuesta correcta puede quedar excluida antes de que se considere, contribuyendo al gran conjunto de espectros no asignados y a un sesgo hacia péptidos abundantes y bien comportados.
Una forma más inteligente de acotar la búsqueda
ProteoRift ofrece una estrategia distinta: en lugar de filtrar candidatos usando solo la masa, aprende a extraer información más rica de cada espectro antes de cualquier búsqueda en la base de datos. El sistema se construye alrededor de una red neuronal profunda con atención que toma como entrada el patrón de picos en un espectro junto con detalles básicos de adquisición. A partir de esto, predice simultáneamente tres propiedades del péptido subyacente: su longitud, cuántas veces fue cortado durante la preparación de la muestra (cortes perdidos), y si lleva alguna modificación. Dado que estas tareas están relacionadas, entrenarlas conjuntamente fomenta que el modelo forme una representación interna robusta de los espectros, mejorando su capacidad de generalizar a datos nuevos.
Convertir las predicciones en búsquedas más rápidas y eficientes
Para poner en práctica estas predicciones, los autores integran ProteoRift en una tubería de extremo a extremo junto con una herramienta desarrollada previamente llamada SpeCollate, que empareja espectros con secuencias de péptidos en un espacio de incrustaciones. Primero, ProteoRift asigna cada espectro a una clase definida por el rango de longitud, el número de cortes perdidos y el estado de modificación. Los péptidos en la base de datos se agrupan de forma similar según sus propiedades conocidas. El motor de búsqueda compara entonces solo espectros con péptidos de la misma clase, en lugar de escanear todos los péptidos con una masa similar. En múltiples conjuntos de datos humanos y del microbioma, este filtrado dirigido reduce teóricamente el espacio de búsqueda de candidatos en más del 90% y ofrece aceleraciones prácticas de aproximadamente 8 a 12 veces en comparación con filtros basados únicamente en la masa, a la vez que recupera números similares de péptidos identificados con confianza. En algunas bases de datos proteogenómicas y metaproteómicas muy grandes, las aceleraciones pueden ser todavía mayores, superando las 40 veces en pruebas específicas.
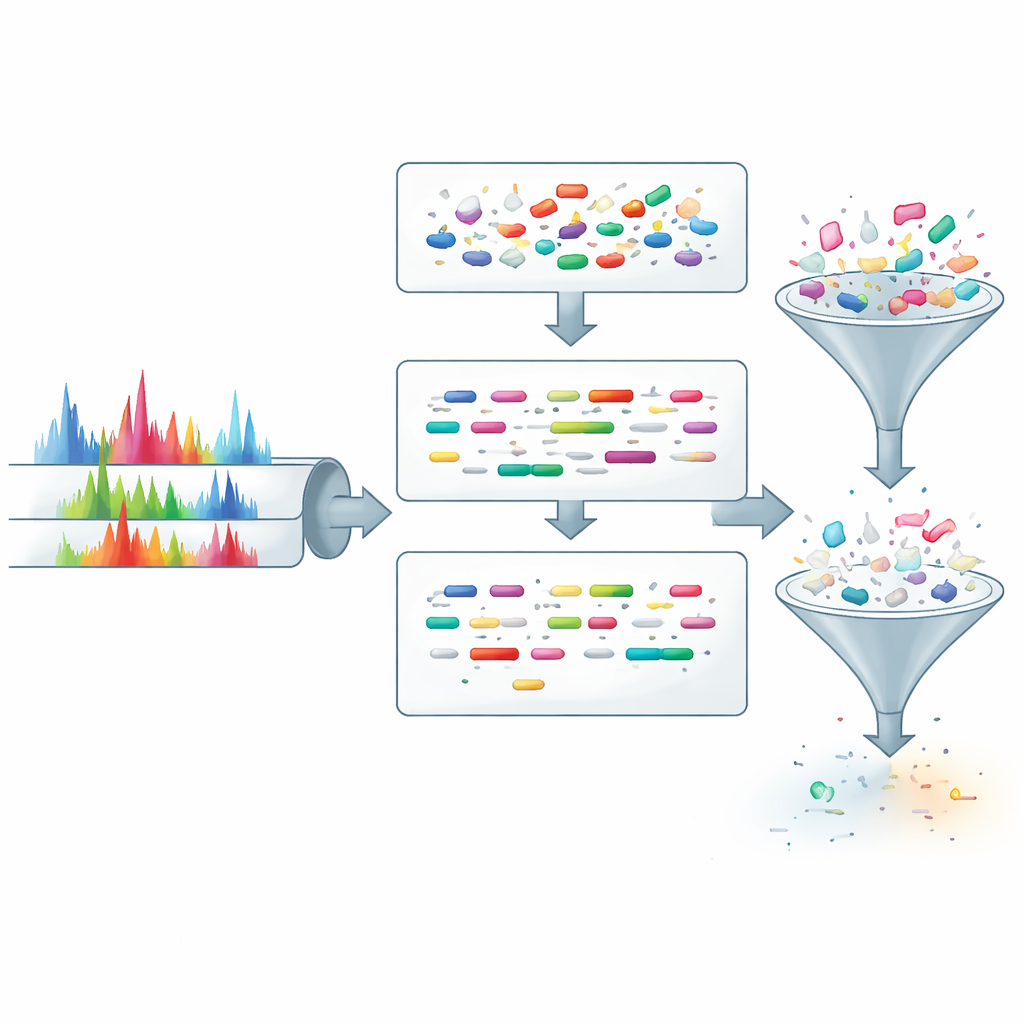
Saber cuándo el modelo podría equivocarse
Porque los sistemas de aprendizaje automático a menudo se perciben como cajas negras, los autores también desarrollan medidas de incertidumbre adaptadas a datos de espectrometría de masas. Investigan cuánto cambia la representación interna de un espectro bajo distorsiones controladas, cuán densamente está rodeado por ejemplos similares de entrenamiento y qué tan bien se preserva la estructura de los datos originales en el espacio aprendido. Estas tres métricas capturan distintos aspectos de la incertidumbre: ruido en las propias mediciones y lagunas en lo que el modelo ha visto durante el entrenamiento. Combinadas, pueden distinguir datos familiares de datos desconocidos con muy alta precisión y ayudar a señalar casos en que la coincidencia de péptido mejor puntuado por el modelo probablemente sea correcta.
Qué significa esto para futuros descubrimientos
En términos cotidianos, ProteoRift funciona como un portero inteligente que mira un espectro y dice: “probablemente esto es un péptido corto, sin modificar, con un corte”, o “esto parece más largo y modificado”, y luego deja entrar solo a los candidatos apropiados para la búsqueda detallada. Al hacerlo, acelera el análisis de forma drástica sin sacrificar gran parte de la precisión, incluso en bases de datos de proteínas complejas o muy grandes. Al mismo tiempo, sus métricas de incertidumbre dan a los investigadores una idea más clara de cuándo confiar en un resultado o cuándo pueden necesitarse más datos o un ajuste fino del modelo. En conjunto, estos avances podrían ayudar a mover la espectrometría de masas más allá de su enfoque actual en proteínas abundantes y bien caracterizadas y abrir nuevas ventanas hacia los péptidos raros y modificados que a menudo contienen las pistas biológicas más interesantes.
Cita: Tariq, U., Shabbir, B. & Saeed, F. End-to-end deep attention-based multitask pipeline for predicting uncertainty-quantified peptide properties from mass spectrometry data. Sci Rep 16, 13331 (2026). https://doi.org/10.1038/s41598-026-43215-2
Palabras clave: proteómica, espectrometría de masas, aprendizaje profundo, identificación de péptidos, estimación de incertidumbre