Clear Sky Science · pt
Modelagem conjunta da heterogeneidade celular e dos efeitos de condição com scPCA em RNA-seq de célula única
Por que isso importa para entender as células
A biologia moderna já consegue ler quais genes estão ativos em milhares de células individuais ao mesmo tempo. Mas quando cientistas comparam células sob diferentes tratamentos, idades ou contextos genéticos, o volume de dados se torna esmagador, e detalhes técnicos podem facilmente ocultar mudanças biológicas reais. Este artigo apresenta uma nova ferramenta analítica, scPCA, que ajuda pesquisadores a separar o que realmente muda nas células sob diferentes condições da diversidade natural que existe entre tipos celulares.
De dados celulares ruidosos a padrões claros
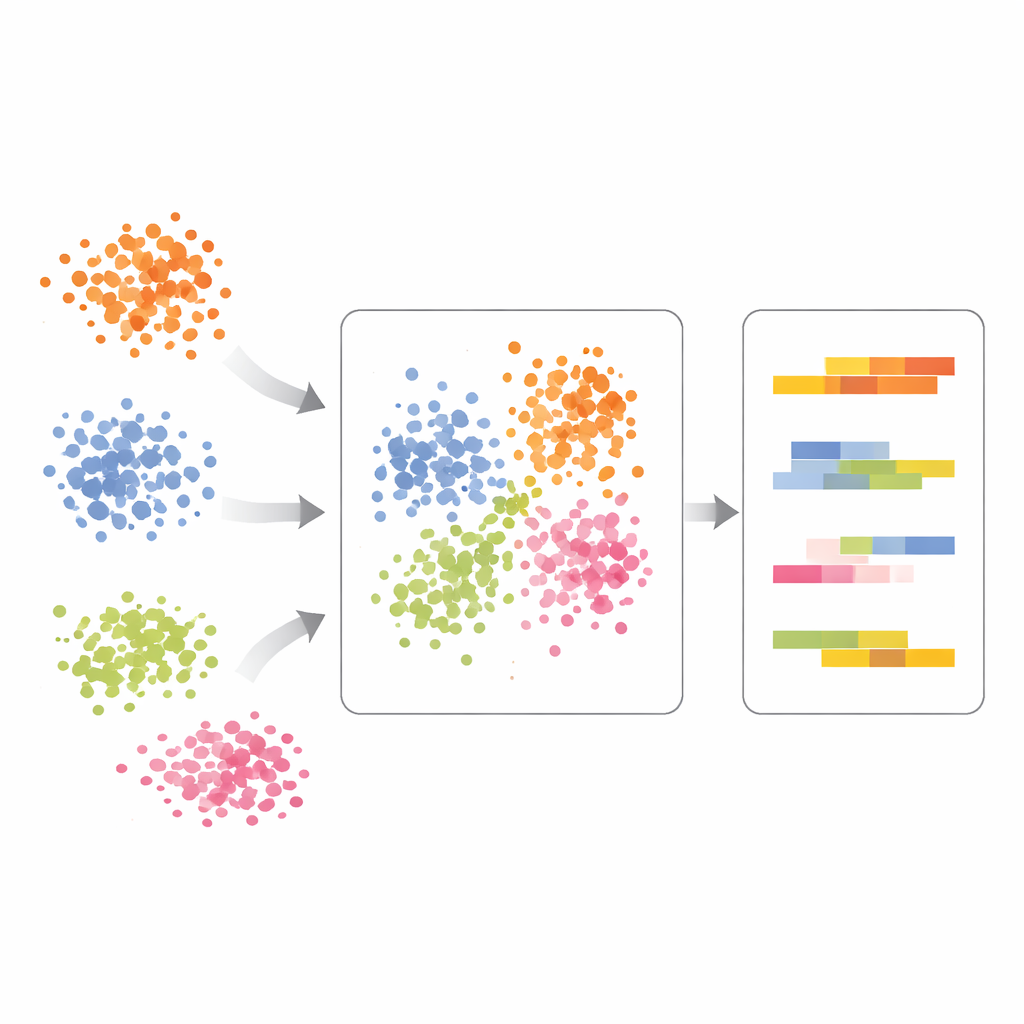
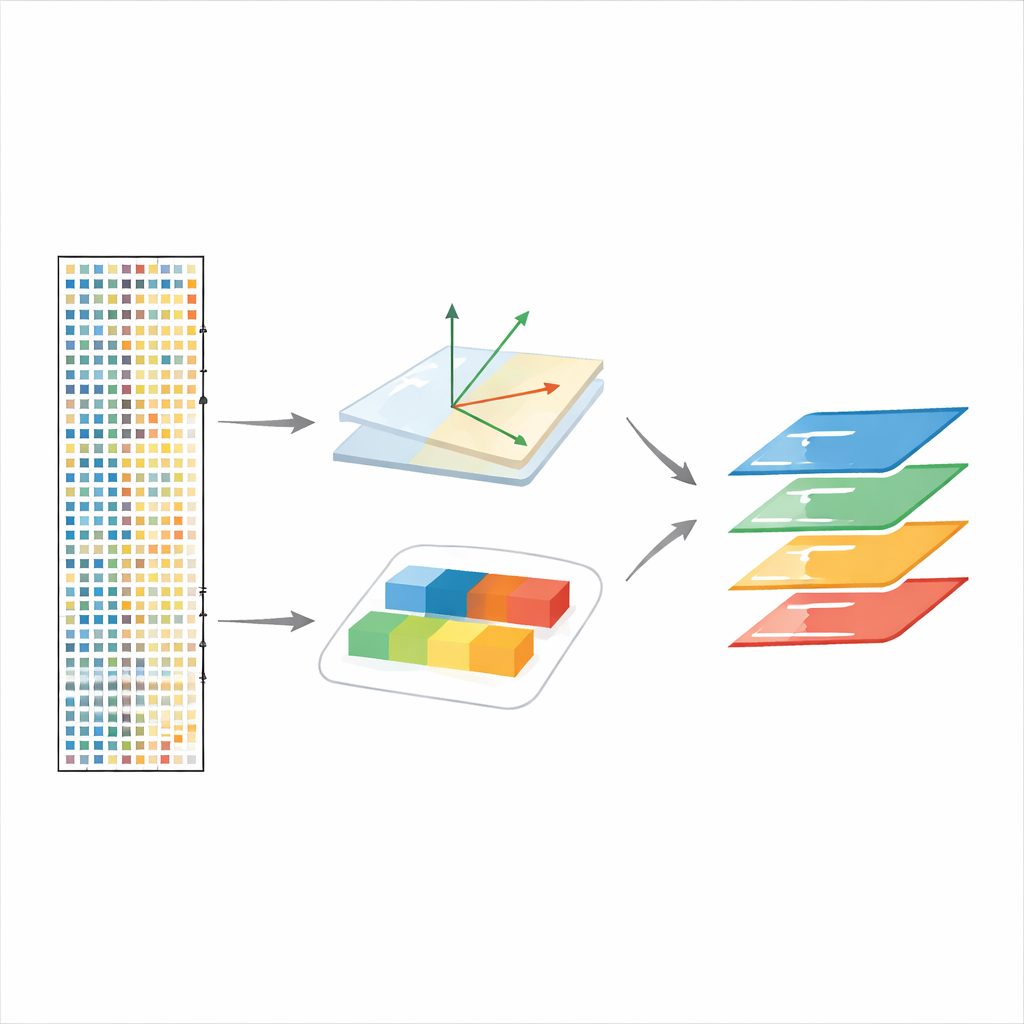
O sequenciamento de RNA de célula única mede a atividade de milhares de genes em cada célula, gerando dados de dimensionalidade extremamente alta. Para interpretar isso, pesquisadores normalmente comprimem os dados com métodos como análise de componentes principais (PCA), que encontra um pequeno conjunto de “eixos” que capturam os principais padrões de variação. No entanto, abordagens tradicionais misturam duas fontes muito diferentes de variação: diferenças inerentes entre tipos celulares e mudanças causadas por um experimento, como um tratamento com medicamento. Os autores argumentam que essa mistura pode induzir análises subsequentes ao erro, como a clusterização de células por tipo ou a busca por efeitos de tratamento.
Uma nova forma de compartilhar estrutura entre condições
O scPCA enfrenta esse problema informando explicitamente ao modelo de fatoração de que condição cada célula veio, e então aprendendo um conjunto separado — porém ligado — de padrões de expressão gênica para cada condição. Em vez de forçar todas as amostras a compartilhar exatamente a mesma estrutura subjacente, o scPCA permite que cada condição tenha sua própria versão de cada componente, levemente deslocada em relação a uma condição de referência designada. Isso preserva um sistema de coordenadas comum para comparar células entre condições, ao mesmo tempo em que captura mudanças sistemáticas de expressão impulsionadas por tratamentos, envelhecimento ou alterações genéticas.
Visualizando efeitos reais de tratamento em células imunes
Os autores demonstram o scPCA em células imunes de pacientes com lúpus, algumas sem tratamento e outras estimuladas com interferon-beta, um sinal imune forte. A análise padrão fez com que as células se agrupassem tanto por tipo celular quanto por tratamento, tornando os resultados difíceis de interpretar. Com scPCA, células do mesmo tipo vindas de diferentes condições se alinharam muito melhor, revelando que o principal eixo de variação ainda refletia linhagens de células imunes em vez do tratamento isoladamente. Só após levar em conta o tipo celular o scPCA destacou deslocamentos induzidos pelo tratamento em genes específicos de células mieloides, incluindo genes ligados à sinalização por interferon e ao processamento proteico alterado dentro da célula. Isso mostrou que o método consegue separar de forma limpa quem são as células de como elas respondem.
Desembaraçando artefatos técnicos e efeitos do envelhecimento
Experimentos frequentemente sofrem com efeitos de lote: diferenças sutis causadas pelo processamento das amostras e não pela biologia. Usando uma mistura de duas linhagens celulares humanas medidas em lotes separados, os autores mostram que a PCA padrão preserva essas diferenças técnicas, enquanto o scPCA pode removê-las em grande parte ao tratar o lote como uma variável condicionante. O método então revela quais genes foram responsáveis pela aparente separação por lote, incluindo marcadores específicos de cada linhagem. Em um exemplo mais complexo, o scPCA foi aplicado a células pulmonares de camundongos jovens e velhos. Ele encontra componentes que se alinham com os principais tipos celulares — como pneumócitos, células ciliadas, macrófagos e células T — e depois aponta mudanças gênicas relacionadas à idade dentro de cada um, incluindo genes de estresse e resposta imune que condizem com a ideia de “inflammaging”.
Acompanhando respostas celulares ao longo do tempo e através de perturbações
O scPCA também lida com experimentos com mais de duas condições, como neurônios no córtex visual de camundongo expostos à luz após um período de escuridão. Ao tratar pontos temporais como diferentes níveis de uma condição, o método recupera ondas precoces e tardias de atividade gênica em vários tipos celulares cerebrais, separando fatores de “resposta precoce” rápidos de programas de “resposta tardia” mais lentos. Em um experimento com zebrafish onde um gene de desenvolvimento importante, chordin, é nocauteado, o scPCA integrou com sucesso embriões apesar de grandes mudanças na composição de tipos celulares e revelou alterações transcricionais consistentes com o padrão corporal alterado, incluindo genes que não foram enfatizados na análise original.
O que isso significa para futuros estudos de célula única
Em termos simples, o scPCA oferece aos pesquisadores uma lente mais clara para observar dados de célula única coletados sob diferentes condições. Ele produz mapas integrados onde células similares se alinham através de tratamentos e destaca quais genes dentro de cada padrão compartilhado estão aumentados ou diminuídos em resposta a um estímulo, envelhecimento ou alteração genética. Embora o método presuma que a estrutura subjacente seja em grande parte compartilhada e seja mais adequado para trabalhos exploratórios que ainda exigem validação adicional, ele oferece uma alternativa mais transparente e interpretável a muitos modelos caixa-preta. Isso deve ajudar cientistas a tirar conclusões mais confiáveis de experimentos de célula única cada vez mais complexos.
Citação: Vöhringer, H. Joint modeling of cellular heterogeneity and condition effects with scPCA in single-cell RNA-seq. Commun Biol 9, 492 (2026). https://doi.org/10.1038/s42003-026-09651-6
Palavras-chave: sequenciamento de RNA de célula única, redução de dimensionalidade, heterogeneidade celular, alterações na expressão gênica, efeitos de lote