Clear Sky Science · pt
Deficiência de SIRT1 promove insuficiência cardíaca relacionada à idade ao aumentar a ferroptose via eixo GATA4-HADHA-GPX4
Por que corações envelhecidos têm dificuldades
À medida que as pessoas vivem mais, mais indivíduos desenvolvem insuficiência cardíaca, uma condição em que o coração deixa de bombear sangue de forma eficaz. Os médicos sabem há muito que o envelhecimento e o excesso de “desgaste” oxidativo prejudicam o coração, mas o tipo exato de dano celular envolvido permaneceu obscuro. Este estudo revela uma forma específica de morte celular dirigida pelo ferro no músculo cardíaco e mapeia uma cadeia de eventos moleculares que pode explicar por que corações mais velhos falham — e como poderíamos retardar ou prevenir esse declínio.
Ferro, ferrugem e corações em falha
Os pesquisadores começaram comparando ratos jovens e idosos. Os animais mais velhos mostraram função de bombeamento enfraquecida, paredes cardíacas mais espessas e rígidas e níveis mais altos no sangue de um hormônio de estresse liberado por corações em dificuldade. No tecido cardíaco deles, a equipe encontrou mais espécies reativas de oxigênio — moléculas altamente reativas que podem danificar lipídios, proteínas e DNA — e fortes indícios de peroxidação lipídica, essencialmente uma “ferrugem” dos componentes gordurosos das membranas celulares. Também observaram acúmulo de ferro e níveis reduzidos de uma enzima protetora chamada GPX4, que normalmente neutraliza oxidantes perigosos derivados de lipídios. Juntas, essas alterações apontam para a ferroptose, um tipo de morte celular descrito recentemente em que ferro e lipídios oxidados se combinam para matar as células.
Demonstrando que a morte celular mediada por ferro importa
Para testar se essa morte celular relacionada ao ferro realmente impulsiona a insuficiência cardíaca em vez de apenas acompanhá‑la, os cientistas manipularam níveis de ferro e a enzima protetora chave GPX4. Fornecer aos ratos idosos uma dieta rica em ferro piorou a função cardíaca e a fibrose e suprimiu ainda mais a GPX4. Em contraste, tratar ratos velhos com ferrostatina‑1, um fármaco que bloqueia especificamente a ferroptose, melhorou o bombeamento cardíaco, reduziu a fibrose e restaurou proteínas de manejo do ferro mais normais. Em camundongos, deletar a GPX4 especificamente nas células do músculo cardíaco agravou os danos e a disfunção relacionados à idade, enquanto aumentar a GPX4 via um vetor de terapia gênica preservou a função em outro modelo de envelhecimento. Esses experimentos sugerem fortemente que a ferroptose não está apenas presente em corações envelhecidos — ela contribui ativamente para seu declínio.
Um ponto fraco metabólico dentro das células cardíacas
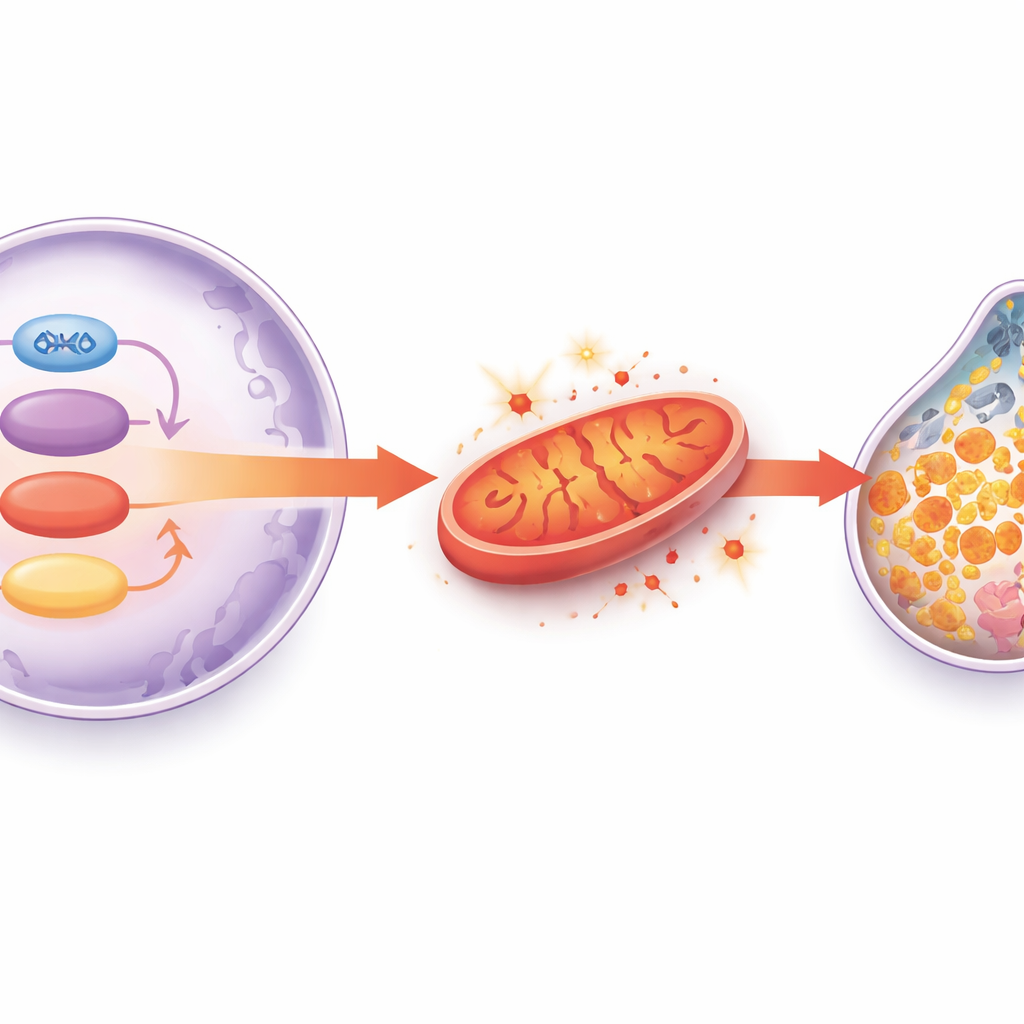
Procurando o que torna as células cardíacas envelhecidas mais vulneráveis à ferroptose, a equipe escaneou milhares de proteínas em corações de ratos e encontrou uma queda marcante em uma enzima mitocondrial chamada HADHA, especialmente quando animais idosos foram expostos a ferro extra. HADHA ajuda as células cardíacas a queimar gorduras de forma eficiente e a manter membranas mitocondriais saudáveis. Quando os cientistas reduziram a HADHA especificamente nas células cardíacas de camundongos jovens, os animais desenvolveram mais fibrose e sinais bioquímicos de ferroptose — alterações que puderam ser revertidas com um inibidor de ferroptose. Em cardiomiócitos isolados, a diminuição de HADHA prejudicou a função mitocondrial, aumentou as espécies reativas de oxigênio, esgotou o antioxidante glutationa e suprimiu ainda mais a GPX4. Restaurar a HADHA teve o efeito oposto, recuperando a produção de energia e as defesas antioxidantes. Isso posiciona a HADHA como um salvaguarda metabólico crucial que impede que as células cardíacas entrem em morte mediada por ferro.
Um interruptor do envelhecimento: SIRT1 e seus parceiros a jusante
A próxima pergunta foi por que a HADHA cai com a idade. Os autores enfocaram a SIRT1, uma proteína há muito associada à longevidade e à proteção contra doenças relacionadas à idade. Em corações envelhecidos, os níveis de SIRT1 caíram junto com a HADHA. Em experimentos celulares, bloquear a SIRT1 reduziu tanto a HADHA quanto a GPX4 e alterou proteínas de manejo do ferro para um perfil mais perigoso. A equipe descobriu que a SIRT1 interage com outra proteína, o fator de transcrição GATA4, que normalmente estimula a produção de HADHA. Quando a atividade de SIRT1 diminui, a capacidade do GATA4 de ativar o gene HADHA fica enfraquecida, fazendo com que os níveis de HADHA caiam e o estresse mitocondrial aumente. Importante: ativar a SIRT1 com resveratrol, uma pequena molécula frequentemente debatida no contexto do “envelhecimento saudável”, ou superexpressar diretamente a SIRT1 em células cardíacas restaurou GATA4 e HADHA, fortaleceu as defesas antioxidantes, reduziu a ferroptose e melhorou a função cardíaca em modelos animais envelhecidos.
O que isso significa para proteger o coração envelhecido
No conjunto, o estudo delineia uma cadeia de eventos: conforme os corações envelhecem, a atividade da SIRT1 diminui, o GATA4 não consegue mais manter os níveis de HADHA, as mitocôndrias falham e liberam espécies reativas de oxigênio, o sistema antioxidante centrado na glutationa e na GPX4 colapsa, e a ferroptose mediada por ferro mata os cardiomiócitos. Essa perda gradual de células e o aumento da fibrose enfraquecem, em última instância, a capacidade de bombeamento do coração. Embora esses resultados provenham de modelos animais e celulares, eles sugerem que fármacos ou estratégias de estilo de vida que preservem a atividade da SIRT1, apoiem a HADHA e o metabolismo mitocondrial, ou bloqueiem diretamente a ferroptose possam um dia ajudar a prevenir ou tratar a insuficiência cardíaca relacionada à idade em humanos.
Citação: Duan, Y., Luo, Y., Han, X. et al. SIRT1 deficiency promotes age-related heart failure through enhancing ferroptosis via GATA4-HADHA-GPX4 axis. Cell Death Dis 17, 343 (2026). https://doi.org/10.1038/s41419-026-08634-z
Palavras-chave: insuficiência cardíaca do envelhecimento, ferroptose, via SIRT1, disfunção mitocondrial, morte de cardiomiócitos