Clear Sky Science · de
SIRT1-Mangel fördert altersbedingte Herzinsuffizienz durch Verstärkung der Ferroptose über die GATA4-HADHA-GPX4-Achse
Warum ältere Herzen Probleme haben
Da die Menschen länger leben, entwickeln immer mehr von uns eine Herzinsuffizienz, einen Zustand, bei dem das Herz nicht mehr effektiv Blut pumpt. Ärztinnen und Ärzte wissen seit Langem, dass Altern und übermäßiger oxidativer "Verschleiß" das Herz schädigen, doch die genaue Art des Zellschadens war lange unklar. Diese Studie enthüllt eine spezifische, eisenabhängige Form des Zelltods im Herzmuskel und kartiert eine Kette molekularer Ereignisse, die erklären könnte, warum ältere Herzen versagen — und wie sich dieser Abbau verlangsamen oder verhindern lässt.
Eisen, Rost und versagende Herzen
Die Forschenden begannen mit einem Vergleich zwischen jungen und alten Ratten. Ältere Tiere zeigten eine geschwächte Pumpfunktion, verdickte und steifere Herzwände sowie höhere Blutspiegel eines Stresshormons, das von belasteten Herzen freigesetzt wird. Im Herzgewebe fanden die Forschenden mehr reaktive Sauerstoffspezies — hochreaktive Moleküle, die Fette, Proteine und DNA schädigen können — sowie starke Anzeichen für Lipidperoxidation, im Grunde ein "Rosten" der fetthaltigen Bestandteile von Zellmembranen. Außerdem beobachteten sie Eisenansammlungen und verringerte Mengen eines schützenden Enzyms namens GPX4, das normalerweise gefährliche, aus Fetten stammende Oxidantien neutralisiert. Zusammengenommen deuten diese Veränderungen auf Ferroptose hin, eine neu beschriebene Form des Zelltods, bei der Eisen und oxidierte Fette zusammen Zellen töten.
Der Beweis, dass eisengetriebener Zelltod relevant ist
Um zu prüfen, ob dieser eisenbezogene Zelltod tatsächlich Herzinsuffizienz antreibt und nicht nur begleitet, manipulierten die Wissenschaftler Eisenwerte und das zentrale Schutzenzym GPX4. Einerseits verschlechterte eine eisenreiche Ernährung bei alten Ratten die Herzfunktion und vermehrte Vernarbung und unterdrückte GPX4 weiter. Andererseits verbesserte die Behandlung alter Ratten mit Ferrostatin-1, einem Wirkstoff, der gezielt Ferroptose blockiert, die Pumpfunktion, reduzierte Narbenbildung und normalisierte teils die Proteine, die am Eisenstoffwechsel beteiligt sind. In Mäusen verschlechterte das gezielte Entfernen von GPX4 in Herzmuskelzellen altersbedingte Schäden und Funktionsstörungen, während eine Steigerung von GPX4 mittels Gentherapiefunktion in einem separaten Alternsmodell die Funktion bewahrte. Diese Experimente legen nahe, dass Ferroptose in alternden Herzen nicht nur vorhanden ist, sondern aktiv zu ihrem Versagen beiträgt.
Eine metabolische Schwachstelle in Herzmuskelzellen
Auf der Suche nach dem Grund, warum gealterte Herzmuskelzellen anfälliger für Ferroptose sind, durchsuchte das Team Tausende von Proteinen in Rattenherzen und fand einen auffälligen Rückgang eines mitochondrialen Enzyms namens HADHA, besonders wenn alte Tiere zusätzlichem Eisen ausgesetzt waren. HADHA hilft Herzmuskelzellen, Fette effizient zu verbrennen und gesunde mitochondriale Membranen zu erhalten. Reduzierten die Forschenden HADHA gezielt in den Herzmuskelzellen junger Mäuse, entwickelten die Tiere mehr Fibrose und biochemische Zeichen der Ferroptose — Veränderungen, die sich mit einem Ferroptose-Inhibitor rückgängig machen ließen. In isolierten Herzzellen störte eine Verringerung von HADHA die mitochondriale Funktion, erhöhte reaktive Sauerstoffspezies, erschöpfte das Antioxidans Glutathion und unterdrückte GPX4 weiter. Die Wiederherstellung von HADHA zeigte den gegenteiligen Effekt, rettete die Energieproduktion und die antioxidativen Abwehrsysteme. Das macht HADHA zu einer entscheidenden metabolischen Schutzfunktion, die Herzmuskelzellen davor bewahrt, in den eisengetriebenen Tod zu kippen.
Ein Altersschalter: SIRT1 und seine nachgeschalteten Partner
Die nächste Frage lautete, warum HADHA mit dem Alter sinkt. Die Autoren konzentrierten sich auf SIRT1, ein Protein, das seit langem mit Lebensspanne und Schutz vor altersbedingten Krankheiten in Verbindung gebracht wird. In alternden Herzen fielen die SIRT1-Spiegel parallel zu HADHA ab. In Zellversuchen führte das Abschalten von SIRT1 zu einer Reduktion sowohl von HADHA als auch von GPX4 und verschob die Eisenhandhabungsproteine in ein gefährlicheres Profil. Das Team entdeckte, dass SIRT1 mit einem anderen Protein interagiert, dem Transkriptionsfaktor GATA4, der normalerweise die HADHA-Produktion steigert. Wenn die SIRT1-Aktivität nachlässt, wird GATA4s Fähigkeit, das HADHA-Gen zu aktivieren, geschwächt, wodurch HADHA-Spiegel sinken und mitochondriale Belastung steigt. Wichtig ist, dass die Aktivierung von SIRT1 mit Resveratrol, einem kleinen Molekül, das oft im Kontext von "gesundem Altern" diskutiert wird, oder das direkte Überexprimieren von SIRT1 in Herzmuskelzellen GATA4 und HADHA wiederherstellte, antioxidative Abwehrkräfte stärkte, Ferroptose reduzierte und die Herzfunktion in Altersmodellen verbesserte.
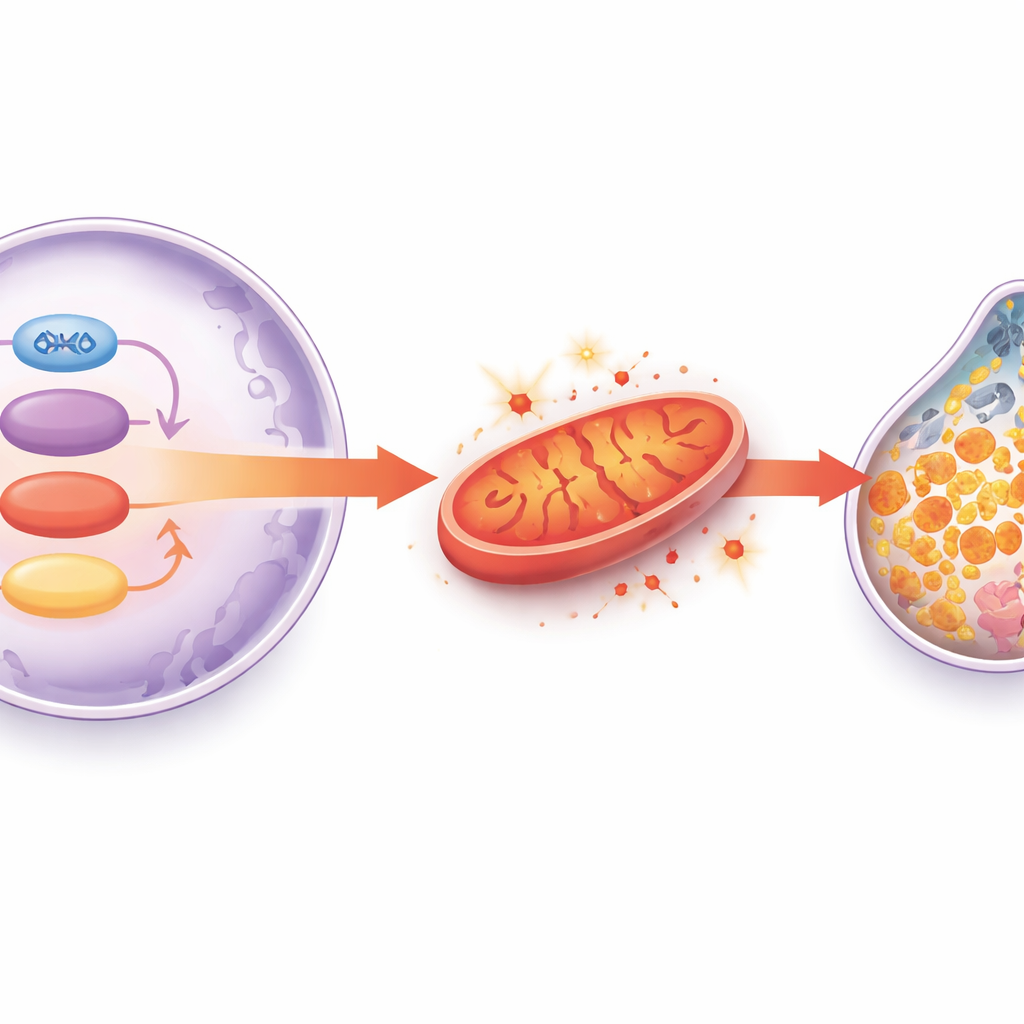
Was das für den Schutz des alternden Herzens bedeutet
Insgesamt skizziert die Studie eine Ereigniskette: Mit zunehmendem Alter sinkt die SIRT1-Aktivität, GATA4 kann HADHA nicht mehr aufrechterhalten, Mitochondrien versagen und setzen reaktive Sauerstoffspezies frei, das auf Glutathion und GPX4 zentrierte antioxidative System bricht zusammen, und eisengetriebene Ferroptose tötet Herzmuskelzellen. Dieser schrittweise Zellverlust und die zunehmende Vernarbung schwächen letztlich die Pumpfunktion des Herzens. Obwohl diese Ergebnisse aus Tier- und Zellmodellen stammen, legen sie nahe, dass Medikamente oder Lebensstilstrategien, die SIRT1-Aktivität erhalten, HADHA und die mitochondriale Stoffwechselversorgung unterstützen oder Ferroptose direkt blockieren, eines Tages helfen könnten, altersbedingte Herzinsuffizienz beim Menschen zu verhindern oder zu behandeln.
Zitation: Duan, Y., Luo, Y., Han, X. et al. SIRT1 deficiency promotes age-related heart failure through enhancing ferroptosis via GATA4-HADHA-GPX4 axis. Cell Death Dis 17, 343 (2026). https://doi.org/10.1038/s41419-026-08634-z
Schlüsselwörter: altersbedingte Herzinsuffizienz, Ferroptose, SIRT1-Signalweg, mitochondriale Dysfunktion, Kardiomyozytensterben