Clear Sky Science · pt
Carcinoma do plexo coróide: estado do campo e direções emergentes
Por que esse raro câncer infantil do cérebro importa
O carcinoma do plexo coróide é um câncer cerebral raro, mas altamente agressivo, que atinge principalmente crianças muito pequenas, frequentemente antes da idade escolar. Como há pouquíssimos pacientes no mundo, existem dados limitados para orientar os médicos, e os tratamentos padrão podem deixar sobreviventes com efeitos colaterais ao longo da vida. Esta revisão reúne o que cientistas e clínicos sabem hoje sobre a doença, como ela surge, como é tratada atualmente e como novos modelos laboratoriais podem finalmente abrir caminho para cuidados mais seguros e mais precisos.
A fábrica de líquido do cérebro e como ela dá errado
No interior de cada cavidade cerebral preenchida por líquido há um tecido fino e franjado chamado plexo coróide. Sua função principal é produzir o líquido cefalorraquidiano, que amortece o cérebro e elimina resíduos. O tecido é formado por células de revestimento especializadas envoltas em pequenos vasos sanguíneos e separadas do líquido por barreiras apertadas. Ao contrário da maioria das células nervosas, as células do plexo coróide ainda podem se dividir lentamente ao longo da vida, o que permite que o tecido se repare após lesões. Essa mesma capacidade de crescimento, entretanto, também o torna vulnerável a se tornar canceroso quando genes chave de controle são danificados.
Um câncer raro com grande impacto nas crianças
O carcinoma do plexo coróide representa apenas cerca de 1% dos tumores cerebrais infantis, mas em bebês com menos de um ano pode corresponder a um quinto de todos os casos. A maioria das crianças é diagnosticada por volta dos três anos, e os tumores geralmente surgem nas cavidades laterais ou posteriores do cérebro. À medida que crescem, frequentemente bloqueiam o fluxo normal do líquido, levando ao acúmulo de pressão denominado hidrocefalia. Famílias e médicos podem notar primeiro o rápido aumento do perímetro craniano, vômitos, problemas oculares, dores de cabeça, convulsões ou mudanças de comportamento. Exames cerebrais mostram massas grandes e irregulares originárias do plexo coróide, mas o diagnóstico definitivo exige exame microscópico e testes moleculares modernos.
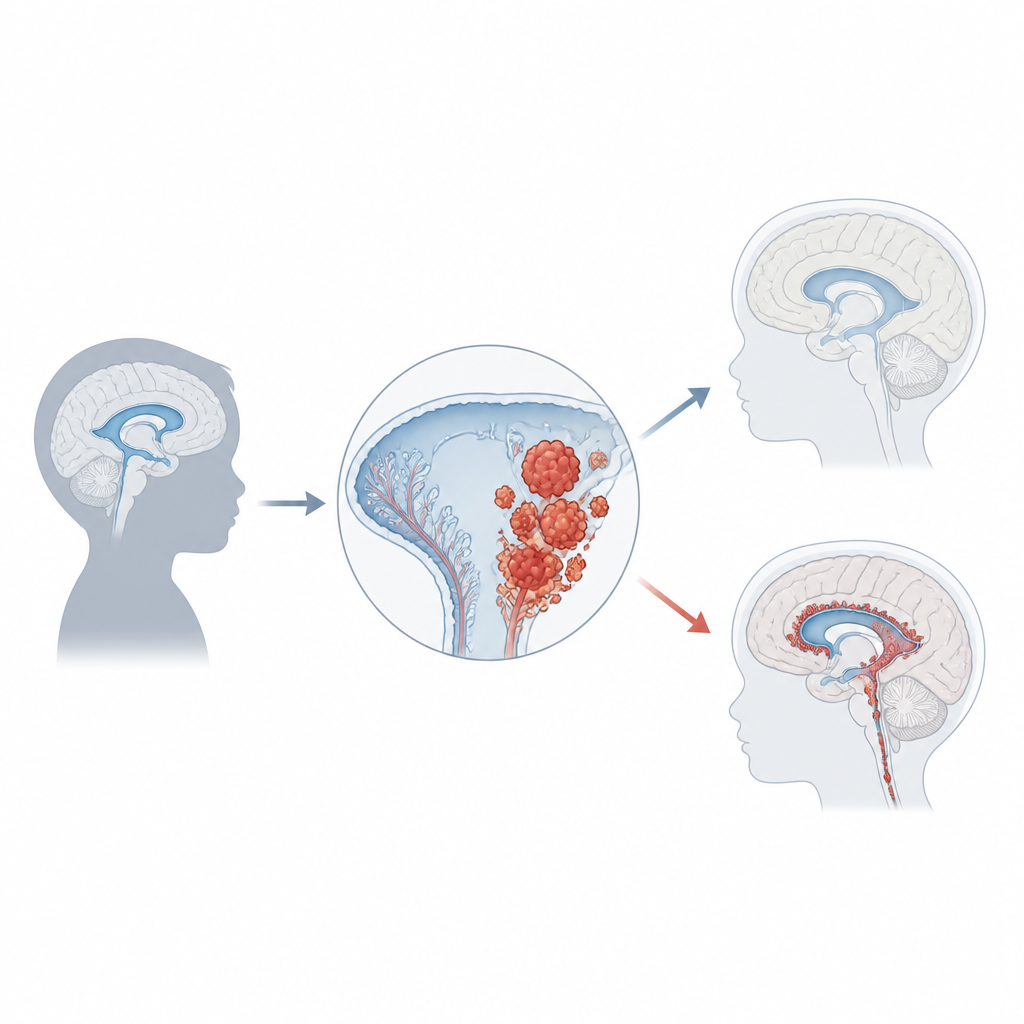
Genes, interruptores e risco tumoral
Uma das pistas mais fortes sobre como esse câncer se forma vem da genética. Cerca da metade dos tumores apresenta alteração em TP53, um gene que normalmente interrompe o crescimento ou aciona a morte celular quando o DNA é danificado. Crianças que herdam mutações em TP53 devido à síndrome de Li‑Fraumeni têm risco especialmente alto, e seus tumores frequentemente acumulam muitas outras alterações no DNA. Esses pacientes tendem a ter prognóstico pior do que aqueles cujo TP53 permanece intacto. Outros genes e vias que impulsionam o crescimento celular ou a resistência à morte, incluindo MYC, Notch e Wnt, também são frequentemente perturbados. Além das mutações no DNA, marcas químicas no DNA que regulam a atividade gênica formam padrões distintos de “metilação”, e um padrão comum nesses tumores se correlaciona com comportamento mais agressivo. Juntas, essas impressões digitais genéticas e epigenéticas começam a classificar os pacientes em subgrupos biologicamente significativos.
Como os médicos tratam hoje
Por enquanto, o fator isolado mais importante associado à sobrevida é o quanto do tumor os cirurgiões conseguem remover com segurança. Crianças cujos tumores são removidos total ou quase totalmente apresentam desfechos muito melhores do que aquelas com grandes remanescentes. Porque o câncer frequentemente se dissemina pelas vias do líquido cerebral, muitas equipes acrescentam quimioterapia e às vezes radiação após a cirurgia. Ainda assim, a radiação pode prejudicar seriamente o cérebro em desenvolvimento, e é particularmente arriscada em crianças com mutações em TP53, que têm maior propensão a desenvolver segundos cânceres induzidos por radiação. Como resultado, os clínicos estão testando combinações de quimioterapias intensivas que visam controlar o tumor poupando ou adiando a radiação, especialmente nos pacientes mais jovens. Resultados iniciais sugerem que certas misturas de drogas podem ajudar, mas o melhor esquema provavelmente depende do perfil molecular de cada tumor.
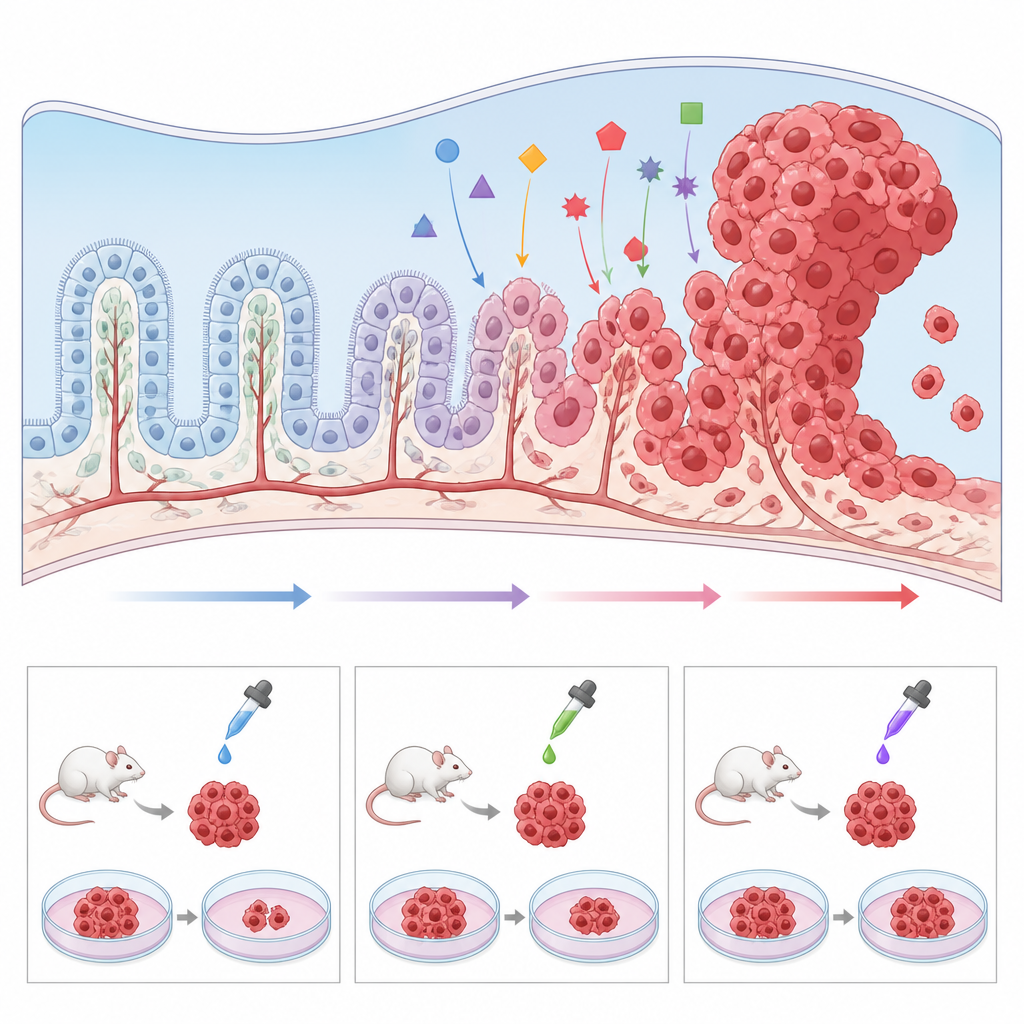
Construindo modelos melhores no laboratório
Como o carcinoma do plexo coróide é tão raro, hospitais individuais veem pouquíssimos casos e amostras tumorais são escassas. Para contornar esse obstáculo, pesquisadores estão desenvolvendo um conjunto de modelos pré‑clínicos. Camundongos geneticamente modificados que carregam as mesmas mutações encontradas em crianças, como perda de TP53 e ativação de MYC, desenvolvem tumores nas mesmas regiões cerebrais e permitem o estudo detalhado de como células normais do plexo coróide são empurradas rumo ao câncer. Tecidos tumorais também podem ser transplantados para camundongos ou zebrafish para testar drogas em um sistema vivo. Paralelamente, cientistas cultivaram linhagens celulares tumorais humanas em placas, assim como “mini‑plexos coróides” a partir de células‑tronco, e demonstraram que ajustar vias como a Wnt pode transformar tecido com aparência saudável em crescimentos semelhantes a tumores. Esses modelos tornam possível realizar triagens de alto rendimento de drogas candidatas e investigar por que alguns tumores resistem à terapia padrão.
Olhando adiante para tratamentos mais pessoais
O artigo conclui que o progresso real contra esse câncer infantil virá de parear o perfil genético e epigenético detalhado do tumor de cada paciente com modelos laboratoriais potentes que reproduzam essas características. Ao aprender quais mutações, mudanças na rede e tipos celulares impulsionam cada caso, os pesquisadores esperam combinar crianças a combinações de drogas mais sob medida e projetar ensaios que evitem o uso indiscriminado de radiação agressiva. Embora o carcinoma do plexo coróide provavelmente continue sendo uma doença rara, essas ferramentas emergentes criam um caminho rumo a maior sobrevida e melhor qualidade de vida para as crianças afetadas.
Citação: Thompson, A., Pescaru, H., Griffin, B. et al. Choroid plexus carcinoma: state of the field and emerging directions. Oncogenesis 15, 21 (2026). https://doi.org/10.1038/s41389-026-00612-6
Palavras-chave: carcinoma do plexo coróide, tumor cerebral pediátrico, mutação TP53, modelos de câncer cerebral, oncologia de precisão