Clear Sky Science · it
Carcinoma del plesso coroideo: stato dell’arte e direzioni emergenti
Perché questo raro tumore cerebrale infantile è importante
Il carcinoma del plesso coroideo è un tumore cerebrale raro ma altamente aggressivo che colpisce prevalentemente bambini molto piccoli, spesso prima dell’età scolare. Poiché i pazienti sono così pochi a livello mondiale, i dati a disposizione dei medici sono limitati e i trattamenti standard possono lasciare i sopravvissuti con effetti collaterali permanenti. Questa rassegna riunisce ciò che scienziati e clinici sanno oggi sulla malattia, su come insorga, su come viene trattata attualmente e su come nuovi modelli di laboratorio possano infine aprire la strada a cure più sicure e più mirate.
La “fabbrica” del liquido cerebrale e come può andare storta
Nelle profondità degli spazi pieni di liquido del cervello si trova un sottile tessuto frastagliato chiamato plesso coroideo. Il suo compito principale è produrre il liquido cerebrospinale, che ammortizza il cervello e rimuove i rifiuti. Il tessuto è costituito da cellule di rivestimento specializzate avvolte intorno a piccoli vasi sanguigni e separate dal fluido da barriere strette. Diversamente dalla maggior parte delle cellule nervose, le cellule del plesso coroideo possono ancora dividersi lentamente per tutta la vita, il che permette al tessuto di ripararsi dopo danni. Questa stessa capacità di crescita, tuttavia, lo rende vulnerabile a trasformarsi in tumore quando geni chiave del controllo vengono danneggiati.
Un cancro raro con grande impatto sui bambini
Il carcinoma del plesso coroideo rappresenta solo circa l’1% dei tumori cerebrali infantili, ma nei bambini sotto un anno può arrivare a costituire un quinto dei casi. La maggior parte dei bambini viene diagnosticata intorno ai tre anni, e i tumori si manifestano di solito negli spazi fluidi laterali o posteriori del cervello. Man mano che crescono, spesso bloccano il normale flusso del liquido, causando un accumulo di pressione chiamato idrocefalo. Famiglie e medici possono notare per primi una rapida crescita della testa, vomito, problemi oculari, mal di testa, convulsioni o cambiamenti comportamentali. Le immagini cerebrali mostrano masse grandi e irregolari che sporgono dal plesso coroideo, ma una diagnosi certa richiede l’esame microscopico e test molecolari moderni.
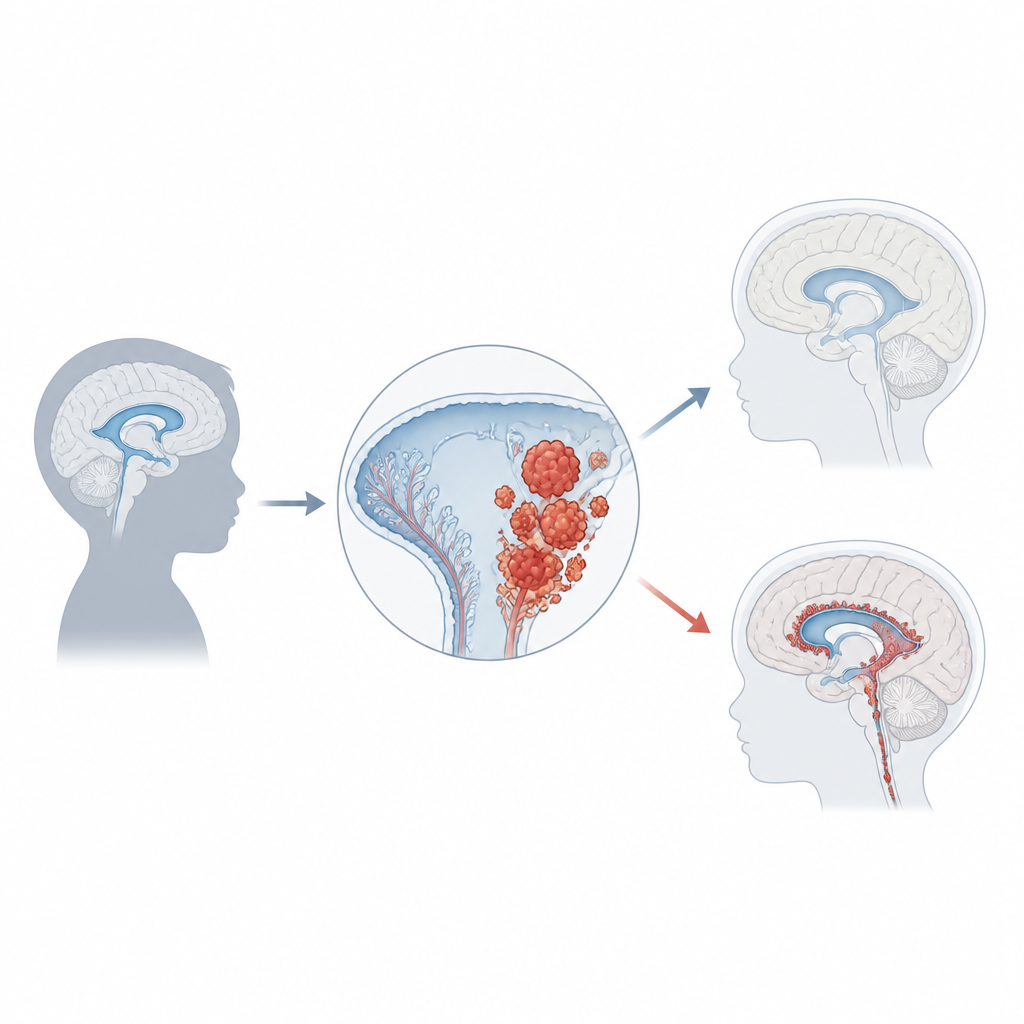
Geni, interruttori e rischio tumorale
Uno degli indizi più solidi su come si formi questo tumore viene dalla genetica. Circa la metà dei tumori presenta danni al gene TP53, un gene che normalmente arresta la crescita o induce la morte cellulare quando il DNA è danneggiato. I bambini che ereditano mutazioni di TP53 tramite la sindrome di Li‑Fraumeni sono particolarmente a rischio, e i loro tumori spesso mostrano molte altre alterazioni del DNA. Questi pazienti tendono ad avere prognosi peggiori rispetto a quelli il cui TP53 resta intatto. Anche altri geni e vie che spingono le cellule a crescere o a resistere alla morte, inclusi MYC, Notch e Wnt, risultano frequentemente disturbati. Oltre alle mutazioni del DNA, etichette chimiche sul DNA che regolano l’attività genica formano distinti modelli di “metilazione”, e un modello comune in questi tumori si correla a un comportamento più aggressivo. Insieme, queste impronte genetiche ed epigenetiche stanno cominciando a dividere i pazienti in sottogruppi biologicamente significativi.
Come lo trattano oggi i medici
Per ora, il fattore singolo più importante correlato alla sopravvivenza è quanto del tumore i chirurghi riescono a rimuovere in sicurezza. I bambini i cui tumori sono completamente o quasi completamente asportati stanno molto meglio rispetto a quelli con grandi residui. Poiché il tumore spesso si diffonde attraverso le vie del liquido cerebrale, molti team aggiungono chemioterapia e talvolta radioterapia dopo l’intervento. Tuttavia la radioterapia può danneggiare gravemente il cervello in sviluppo ed è particolarmente rischiosa nei bambini con mutazioni TP53, che sono più suscettibili a secondi tumori indotti dalle radiazioni. Per questo motivo i clinici stanno testando combinazioni di chemioterapie intensive che mirano a controllare il tumore risparmiando o ritardando la radioterapia, soprattutto nei pazienti più piccoli. I risultati iniziali suggeriscono che alcune combinazioni di farmaci possono essere d’aiuto, ma la migliore terapia dipenderà probabilmente dal profilo molecolare del singolo tumore.
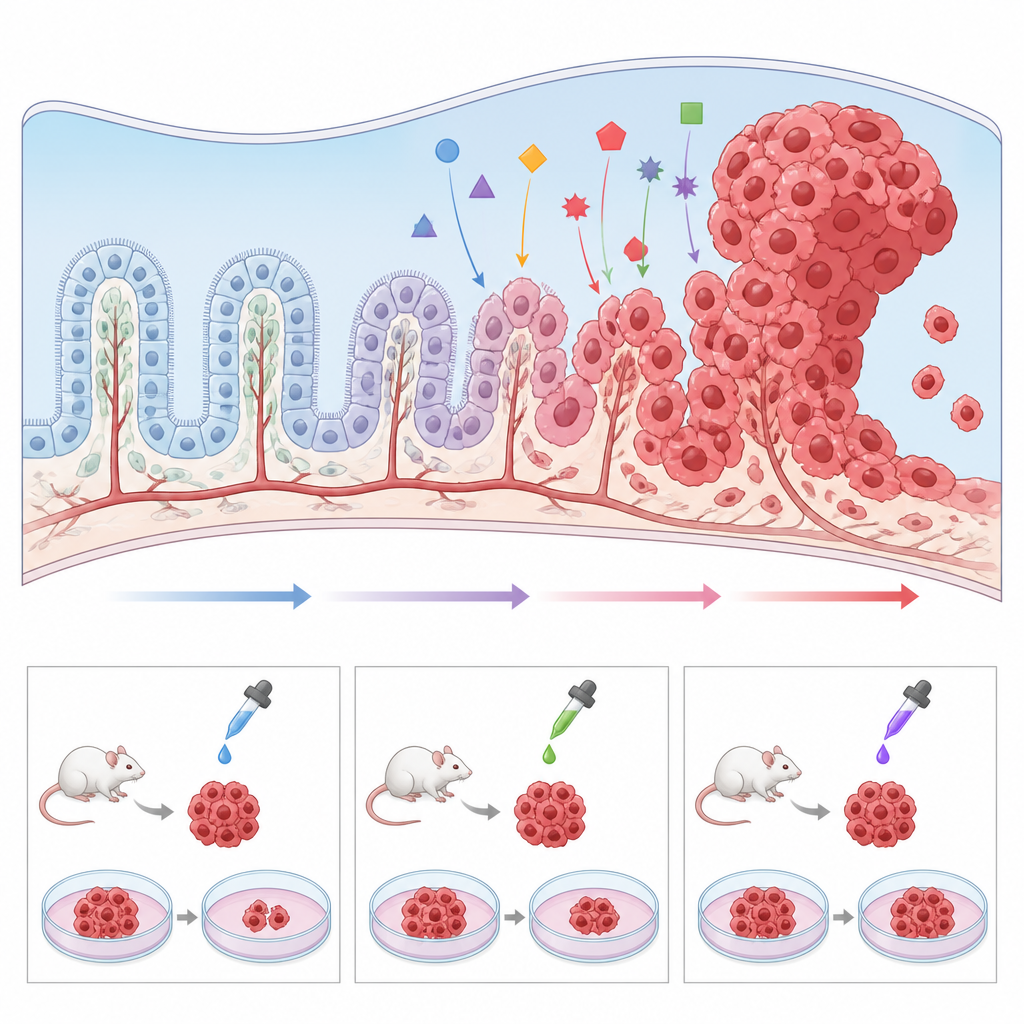
Costruire modelli migliori in laboratorio
Poiché il carcinoma del plesso coroideo è così raro, i singoli ospedali vedono pochissimi casi e i campioni tumorali sono scarsi. Per superare questo ostacolo, i ricercatori stanno sviluppando una serie di modelli preclinici. Topi geneticamente modificati che portano le stesse mutazioni trovate nei bambini, come la perdita di TP53 e l’attivazione di MYC, sviluppano tumori nelle stesse regioni cerebrali e permettono di studiare con attenzione come le cellule normali del plesso coroideo vengano spinte verso il cancro. Il tessuto tumorale può anche essere trapiantato in topi o zebrafish per testare farmaci in un sistema vivente. In parallelo, gli scienziati hanno coltivato linee cellulari tumorali umane in piastre, così come “mini plessi coroidei” da cellule staminali, dimostrando che la modulazione di vie come Wnt può trasformare tessuto con caratteristiche sane in crescite simili a tumori. Questi modelli rendono possibile effettuare screening ad alto rendimento di farmaci candidati ed esplorare perché alcuni tumori resistono alle terapie standard.
Guardando avanti a trattamenti più personalizzati
L’articolo conclude che i veri progressi contro questo tumore infantile deriveranno dall’abbinare un profilo genetico ed epigenetico accurato di ogni tumore con potenti modelli di laboratorio che riflettano quelle caratteristiche. Imparando quali mutazioni, cambiamenti nei circuiti e tipi cellulari guidano ciascun caso, i ricercatori sperano di associare i bambini a combinazioni di farmaci più mirate e di progettare studi clinici che evitino l’uso indiscriminato di radioterapia dannosa. Sebbene il carcinoma del plesso coroideo rimarrà probabilmente una malattia rara, questi strumenti emergenti creano un percorso verso una sopravvivenza più lunga e una migliore qualità di vita per i bambini colpiti.
Citazione: Thompson, A., Pescaru, H., Griffin, B. et al. Choroid plexus carcinoma: state of the field and emerging directions. Oncogenesis 15, 21 (2026). https://doi.org/10.1038/s41389-026-00612-6
Parole chiave: carcinoma del plesso coroideo, tumore cerebrale pediatrico, mutazione TP53, modelli di cancro cerebrale, oncologia di precisione