Clear Sky Science · es
Carcinoma del plexo coroideo: estado del campo y direcciones emergentes
Por qué importa este raro cáncer infantil del cerebro
El carcinoma del plexo coroideo es un cáncer cerebral raro pero muy agresivo que afecta principalmente a niños muy pequeños, a menudo antes de la edad escolar. Debido a que existen muy pocos pacientes en todo el mundo, hay datos limitados para guiar a los médicos, y los tratamientos estándar pueden dejar a los supervivientes con efectos secundarios de por vida. Esta revisión recopila lo que científicos y clínicos saben hoy sobre la enfermedad, cómo se origina, cómo se trata actualmente y cómo los nuevos modelos de laboratorio pueden finalmente abrir la puerta a cuidados más seguros y precisos.
La “fábrica” de líquido del cerebro y cómo falla
En lo profundo de cada una de las cavidades llenas de líquido del cerebro se encuentra un tejido delgado y fruncido llamado plexo coroideo. Su función principal es producir el líquido cefalorraquídeo, que amortigua el cerebro y elimina residuos. El tejido está compuesto por células epiteliales especializadas que rodean diminutos vasos sanguíneos y están separadas del líquido por barreras estrechas. A diferencia de la mayoría de las neuronas, las células del plexo coroideo pueden dividirse lentamente a lo largo de la vida, lo que permite que el tejido se repare tras una lesión. Esa misma capacidad de crecimiento, sin embargo, también lo hace vulnerable a convertirse en cáncer cuando se dañan genes clave de control.
Un cáncer raro con un gran impacto en la infancia
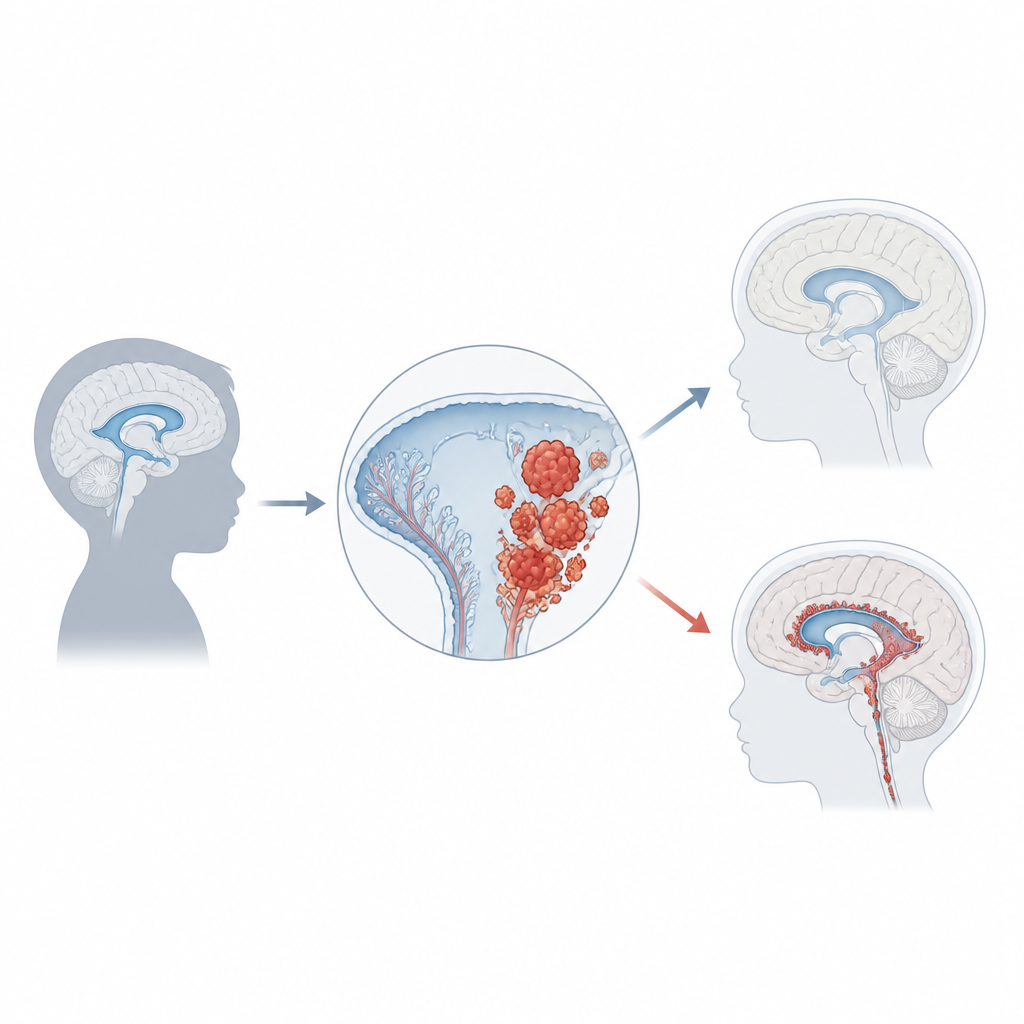
El carcinoma del plexo coroideo representa solo alrededor del 1 por ciento de los tumores cerebrales infantiles, pero en bebés menores de un año puede constituir una quinta parte de los casos. La mayoría de los niños son diagnosticados hacia los tres años, y los tumores suelen originarse en los espacios laterales o posteriores llenos de líquido del cerebro. A medida que crecen, con frecuencia obstruyen el flujo normal del líquido, provocando una acumulación de presión llamada hidrocefalia. Las familias y los médicos pueden notar primero un rápido aumento del tamaño de la cabeza, vómitos, problemas oculares, dolores de cabeza, convulsiones o cambios en el comportamiento. Las exploraciones cerebrales muestran masas grandes e irregulares que brotan del plexo coroideo, pero un diagnóstico firme requiere examen microscópico y pruebas moleculares modernas.
Genes, interruptores y riesgo tumoral
Una de las pistas más sólidas sobre cómo se forma este cáncer proviene de la genética. Aproximadamente la mitad de los tumores presentan alteraciones en TP53, un gen que normalmente detiene el crecimiento o desencadena la muerte celular cuando el ADN está dañado. Los niños que heredan mutaciones en TP53 a través del síndrome de Li‑Fraumeni tienen un riesgo especialmente alto, y sus tumores suelen contener muchos otros cambios en el ADN. Estos pacientes tienden a tener peor pronóstico que aquellos cuyo TP53 permanece intacto. Otros genes y vías que impulsan el crecimiento celular o la resistencia a la muerte, incluidas MYC, Notch y Wnt, también se ven frecuentemente alterados. Además de las mutaciones en el ADN, marcas químicas sobre el ADN que regulan la actividad génica forman patrones distintivos de “metilación”, y un patrón común en estos tumores se correlaciona con un comportamiento más agresivo. En conjunto, estas huellas genéticas y epigenéticas empiezan a clasificar a los pacientes en subgrupos biológicamente relevantes.
Cómo lo tratan hoy los médicos
Por ahora, el factor único más importante ligado a la supervivencia es la cantidad de tumor que los cirujanos pueden extirpar de forma segura. Los niños cuyos tumores se resecan total o casi totalmente tienen un pronóstico mucho mejor que aquellos con grandes restos. Debido a que el cáncer a menudo se disemina por las vías de líquido del cerebro, muchos equipos añaden quimioterapia y, en ocasiones, radioterapia después de la cirugía. Sin embargo, la radiación puede dañar seriamente el cerebro en desarrollo y es particularmente arriesgada en niños con mutaciones en TP53, que son más propensos a desarrollar segundos cánceres inducidos por radiación. Como resultado, los clínicos están probando combinaciones de quimioterapia intensiva que buscan controlar el tumor mientras se evita o retrasa la radiación, especialmente en los pacientes más jóvenes. Los resultados iniciales sugieren que ciertas mezclas de fármacos pueden ayudar, pero el mejor régimen probablemente dependa del perfil molecular de cada tumor.
Construir mejores modelos en el laboratorio
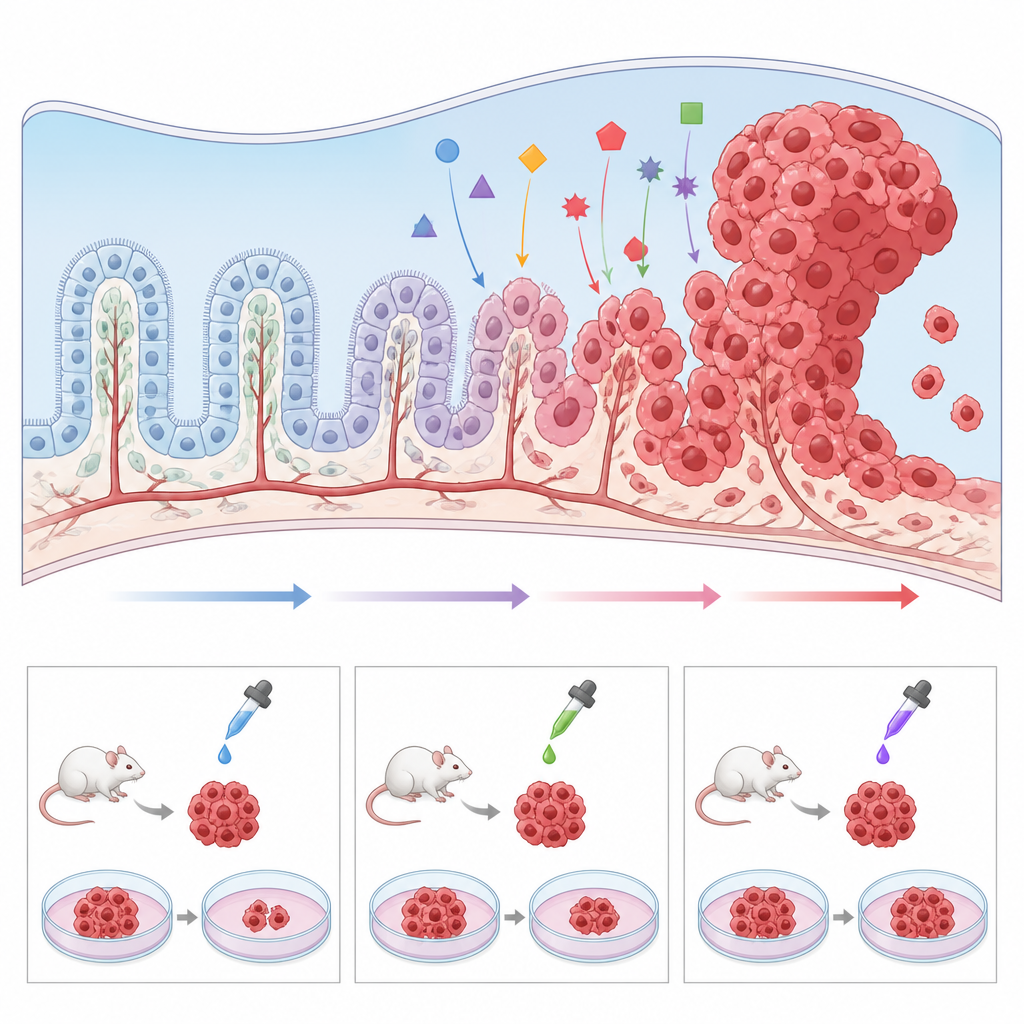
Dado que el carcinoma del plexo coroideo es tan raro, los hospitales individuales ven muy pocos casos y las muestras tumorales son escasas. Para sortear este obstáculo, los investigadores están desarrollando una serie de modelos preclínicos. Ratones genéticamente modificados que portan las mismas mutaciones encontradas en niños, como la pérdida de TP53 y la activación de MYC, desarrollan tumores en las mismas regiones cerebrales y permiten estudiar con detalle cómo las células normales del plexo coroideo son empujadas hacia el cáncer. El tejido tumoral también puede trasplantarse a ratones o pez cebra para probar fármacos en un sistema vivo. Paralelamente, los científicos han cultivado líneas celulares humanas tumorales en platos, así como “mini plexos coroideos” a partir de células madre, y han demostrado que modificar vías como Wnt puede transformar tejido de aspecto sano en crecimiento similar al tumoral. Estos modelos permiten realizar cribados de alto rendimiento de fármacos candidatos y explorar por qué algunos tumores resisten la terapia estándar.
Mirando hacia tratamientos más personalizados
El artículo concluye que el progreso real contra este cáncer infantil vendrá de emparejar un perfil genético y epigenético cuidadoso del tumor de cada paciente con modelos de laboratorio potentes que reflejen esas características. Al identificar qué mutaciones, cambios en el cableado molecular y tipos celulares impulsan cada caso, los investigadores esperan asignar a los niños combinaciones de fármacos más adaptadas y diseñar ensayos que eviten el uso generalizado de radiación dañina. Aunque el carcinoma del plexo coroideo probablemente seguirá siendo una enfermedad rara, estas herramientas emergentes crean un camino hacia una mayor supervivencia y mejor calidad de vida para los niños afectados.
Cita: Thompson, A., Pescaru, H., Griffin, B. et al. Choroid plexus carcinoma: state of the field and emerging directions. Oncogenesis 15, 21 (2026). https://doi.org/10.1038/s41389-026-00612-6
Palabras clave: carcinoma del plexo coroideo, tumor cerebral pediátrico, mutación TP53, modelos de cáncer cerebral, oncología de precisión