Clear Sky Science · nl
Choroïd plexuscarcinoom: stand van zaken en nieuwe richtingen
Waarom deze zeldzame kinderhersenkanker ertoe doet
Choroïd plexuscarcinoom is een zeldzame maar zeer agressieve hersenkanker die vooral zeer jonge kinderen treft, vaak nog voor ze naar de kleuterschool gaan. Omdat er wereldwijd zo weinig patiënten zijn, zijn de gegevens voor artsen beperkt en kunnen standaardbehandelingen overlevenden levenslange bijwerkingen bezorgen. Deze review brengt samen wat wetenschappers en clinici inmiddels weten over de ziekte, hoe ze ontstaat, hoe ze tegenwoordig wordt behandeld en hoe nieuwe laboratoriummodellen mogelijk de deur openen naar veiliger, preciezer zorg.
De vloeistoffabriek van de hersenen en hoe het misgaat
Diep in de met vocht gevulde ruimten van de hersenen ligt een dun, gegolfd weefsel dat het choroïd plexus wordt genoemd. De belangrijkste taak is het produceren van cerebrospinale vloeistof, die de hersenen beschermt en afvalstoffen afvoert. Het weefsel bestaat uit gespecialiseerde bekercellen rondom kleine bloedvaatjes en wordt van het vocht gescheiden door strakke barrières. In tegenstelling tot de meeste zenuwcellen kunnen choroïd plexuscellen langzaam blijven delen gedurende het leven, wat het weefsel in staat stelt zichzelf te herstellen na schade. Diezelfde groeicapaciteit maakt het weefsel echter ook kwetsbaar voor kanker als belangrijke regelgenen beschadigd raken.
Een zeldzame kanker met grote impact op kinderen
Choroïd plexuscarcinoom is verantwoordelijk voor slechts ongeveer 1 procent van de kinderlijke hersentumoren, maar bij zuigelingen onder één jaar kan het tot een vijfde van de gevallen uitmaken. De meeste kinderen worden rond de leeftijd van drie jaar gediagnosticeerd en de tumoren ontstaan doorgaans in de zij- of achterste vochtruimten van de hersenen. Als ze groeien, blokkeren ze vaak de normale vloeistofstroom, wat leidt tot drukopbouw die hydrocefalus wordt genoemd. Families en artsen merken mogelijk eerst snel hoofdgroei, braken, oogproblemen, hoofdpijn, aanvallen of gedragsveranderingen op. Hersenonderzoeken tonen grote, onregelmatige massa’s die uit het choroïd plexus ontspringen, maar een definitieve diagnose vereist microscopisch onderzoek en moderne moleculaire tests.
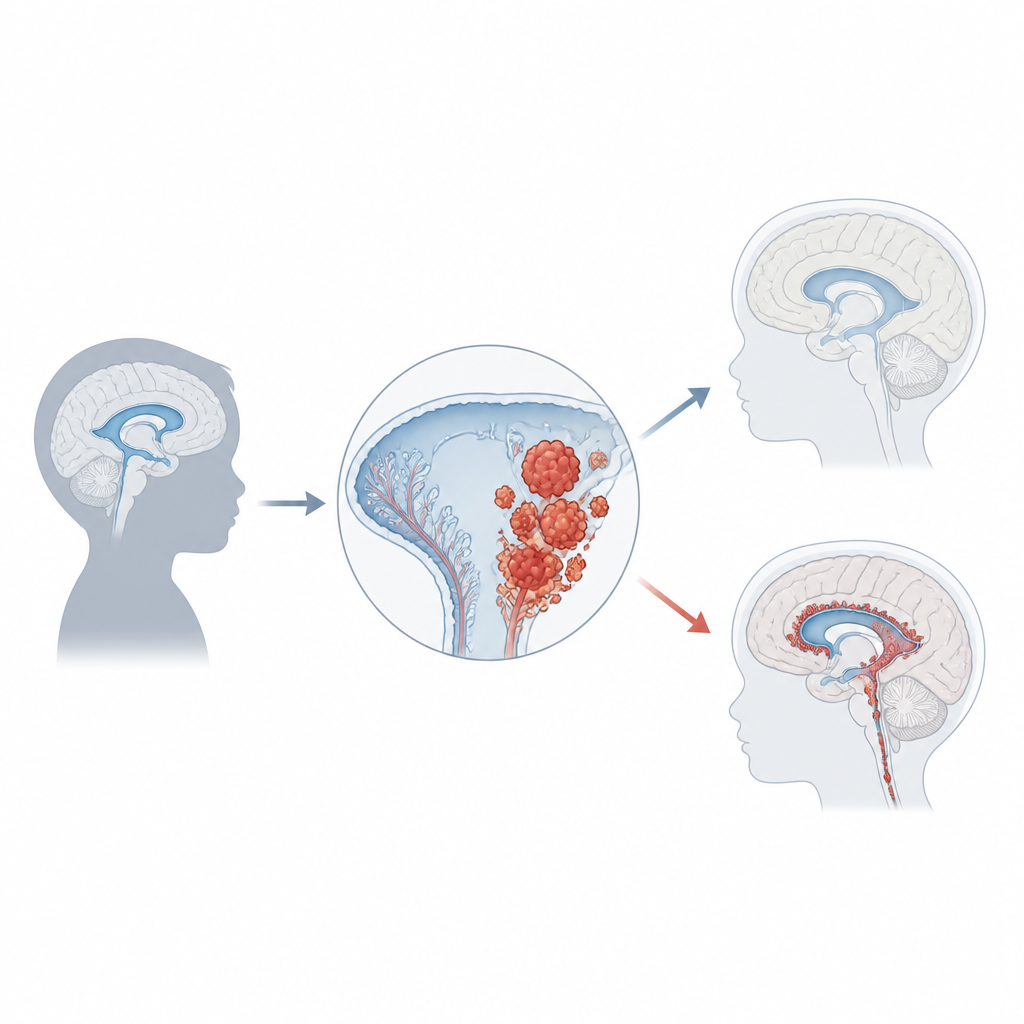
Genen, schakelaars en tumorrisico
Een van de meest aanwijzende factoren voor hoe deze kanker ontstaat komt uit de genetica. Ongeveer de helft van de tumoren bevat schade in TP53, een gen dat normaal de groei stillegt of celdood induceert wanneer DNA beschadigd is. Kinderen die TP53-mutaties erven via het Li-Fraumeni-syndroom lopen een bijzonder hoog risico en hun tumoren bevatten vaak veel andere DNA-veranderingen. Deze patiënten doen het doorgaans slechter dan degenen met intacte TP53. Andere genen en routes die cellen aanzetten tot delen of resistentie tegen celdood, waaronder MYC, Notch en Wnt, zijn ook vaak verstoord. Naast DNA-mutaties vormen chemische labels op het DNA die genactiviteit reguleren—zogeheten methylatiepatronen—karakteristieke profielen; één veelvoorkomend patroon bij deze tumoren correleert met agressiever gedrag. Samen beginnen deze genetische en epigenetische vingerafdrukken patiënten in biologisch relevante subgroepen te verdelen.
Hoe artsen het tegenwoordig behandelen
Vooralsnog is de belangrijkste factor die samenhangt met overleving hoe groot een gedeelte van de tumor chirurgen veilig kunnen wegnemen. Kinderen wier tumoren volledig of nagenoeg volledig worden verwijderd, hebben aanzienlijk betere vooruitzichten dan kinderen met grote resttumoren. Omdat de kanker zich vaak via de vochtpaden van de hersenen verspreidt, voegen veel teams na de operatie chemotherapie en soms bestraling toe. Bestraling kan echter de zich ontwikkelende hersenen ernstig beschadigen en is bijzonder risicovol bij kinderen met TP53-mutaties, die gevoelig zijn voor door bestraling geïnduceerde secundaire kankers. Daarom testen clinici combinaties van intensieve chemotherapie die proberen de tumor te beheersen terwijl bestraling wordt uitgespaard of uitgesteld, vooral bij de jongste patiënten. Vroege resultaten suggereren dat bepaalde geneesmiddelmengsels kunnen helpen, maar het beste regime hangt waarschijnlijk af van het moleculaire profiel van elke tumor.
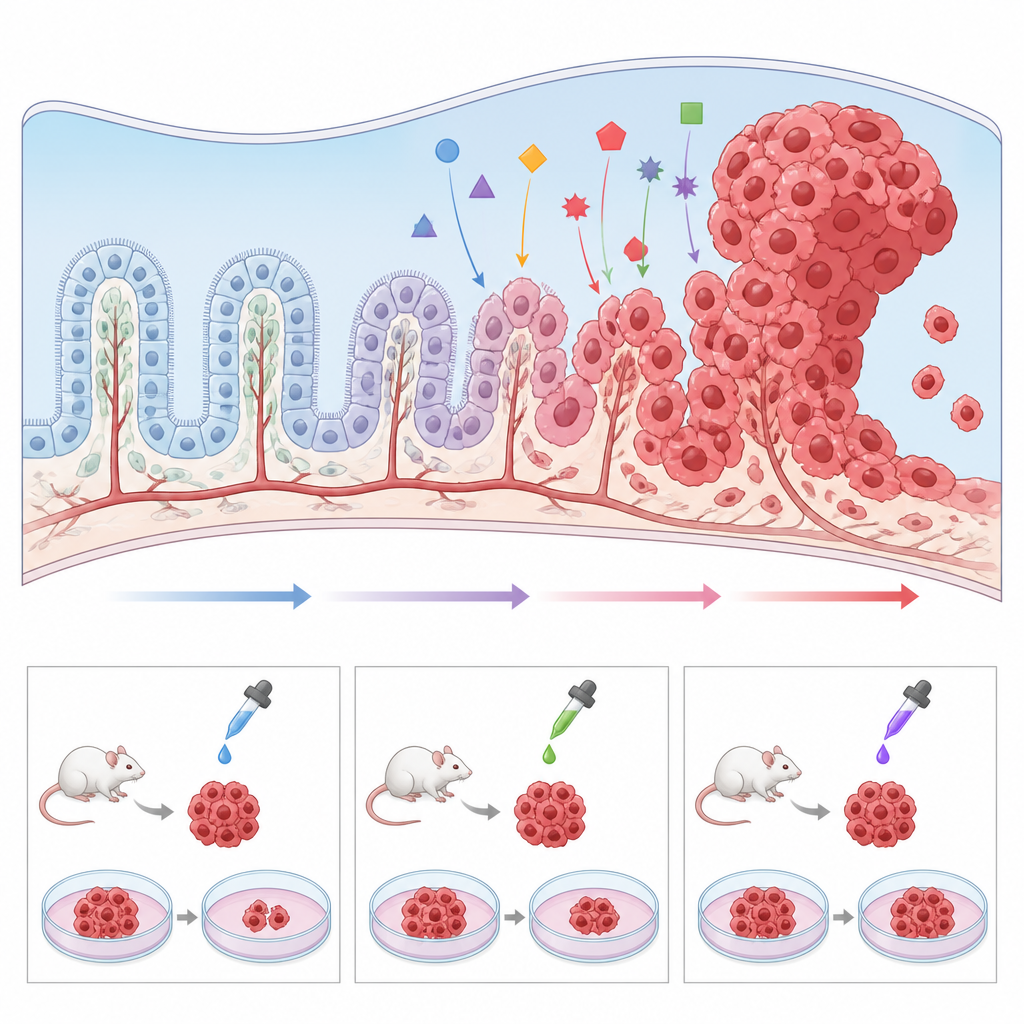
Betere modellen bouwen in het laboratorium
Aangezien choroïd plexuscarcinoom zo zeldzaam is, zien individuele ziekenhuizen maar weinig gevallen en zijn tumorbiopten schaars. Om dit obstakel te omzeilen, ontwikkelen onderzoekers een reeks preklinische modellen. Genetisch gemodificeerde muizen die dezelfde mutaties dragen als bij kinderen, zoals verlies van TP53 en activatie van MYC, ontwikkelen tumoren in dezelfde hersengebieden en maken nauwkeurig onderzoek mogelijk naar hoe normale choroïd plexuscellen naar kanker worden geduwd. Tumorweefsel kan ook worden getransplanteerd in muizen of zebravissen om geneesmiddelen in een levend systeem te testen. Parallel daaraan hebben wetenschappers menselijke tumorcelijnen in kweek gebracht, evenals "mini-choroïd plexus" structuren uit stamcellen, en laten ze zien dat het bijsturen van routes zoals Wnt gezond-achtig weefsel in tumorachtig groeien kan transformeren. Deze modellen maken high-throughput screens van kandidaat-geneesmiddelen mogelijk en helpen te onderzoeken waarom sommige tumoren resistent zijn tegen standaardtherapie.
Vooruitkijken naar meer persoonsgerichte behandeling
Het artikel concludeert dat echte vooruitgang tegen deze kinderhersenkanker zal komen door nauwkeurige genetische en epigenetische profilering van de tumor van elke patiënt te koppelen aan krachtige laboratoriummodellen die die kenmerken nabootsen. Door te leren welke mutaties, netwerkveranderingen en celtypen elke casus aandrijven, hopen onderzoekers kinderen te koppelen aan meer op maat gemaakte geneesmiddelpakketten en trials te ontwerpen die het algeheel gebruik van zware bestraling vermijden. Hoewel choroïd plexuscarcinoom waarschijnlijk een zeldzame ziekte zal blijven, scheppen deze opkomende hulpmiddelen een pad naar langere overleving en betere levenskwaliteit voor getroffen kinderen.
Bronvermelding: Thompson, A., Pescaru, H., Griffin, B. et al. Choroid plexus carcinoma: state of the field and emerging directions. Oncogenesis 15, 21 (2026). https://doi.org/10.1038/s41389-026-00612-6
Trefwoorden: choroïd plexuscarcinoom, pediatrische hersentumor, TP53-mutatie, hersen-kankermodellen, precisie-oncologie