Clear Sky Science · fr
Carcinome du plexus choroïde : état des lieux et orientations émergentes
Pourquoi ce cancer infantile rare compte
Le carcinome du plexus choroïde est un cancer cérébral rare mais très agressif qui touche principalement de très jeunes enfants, souvent avant l’âge de l’école maternelle. Parce que si peu de patients existent dans le monde, les données disponibles sont limitées pour guider les médecins, et les traitements standards peuvent laisser des survivants avec des effets secondaires à vie. Cette revue rassemble ce que scientifiques et cliniciens savent aujourd’hui sur la maladie, son origine, sa prise en charge actuelle et la manière dont de nouveaux modèles en laboratoire pourraient enfin ouvrir la voie à des soins plus sûrs et plus ciblés.
L’usine à liquide du cerveau et ce qui peut mal tourner
Au cœur des cavités remplies de liquide du cerveau se trouve un tissu mince et froncé appelé plexus choroïde. Sa fonction principale est de produire le liquide cérébro‑spinal, qui amortit le cerveau et élimine les déchets. Le tissu est constitué de cellules épithéliales spécialisées entourant de petits vaisseaux sanguins et séparées du liquide par des barrières serrées. Contrairement à la plupart des neurones, les cellules du plexus choroïde peuvent encore se diviser lentement tout au long de la vie, ce qui permet au tissu de se réparer après une blessure. Cette même capacité de prolifération le rend cependant vulnérable à une transformation cancéreuse lorsque des gènes de contrôle clés sont endommagés.
Un cancer rare avec un lourd impact chez l’enfant
Le carcinome du plexus choroïde représente seulement environ 1 % des tumeurs cérébrales infantiles, mais chez les nourrissons de moins d’un an il peut constituer un cinquième des cas. La plupart des enfants sont diagnostiqués vers l’âge de trois ans, et les tumeurs se développent habituellement dans les cavités latérales ou postérieures du cerveau. En grossissant, elles bloquent souvent l’écoulement normal du liquide, entraînant une pression accrue appelée hydrocéphalie. Les familles et les médecins peuvent d’abord remarquer une croissance rapide du crâne, des vomissements, des troubles oculaires, des maux de tête, des convulsions ou des changements de comportement. Les imageries cérébrales montrent de larges masses irrégulières prenant naissance au niveau du plexus choroïde, mais un diagnostic formel nécessite un examen microscopique et des tests moléculaires modernes.
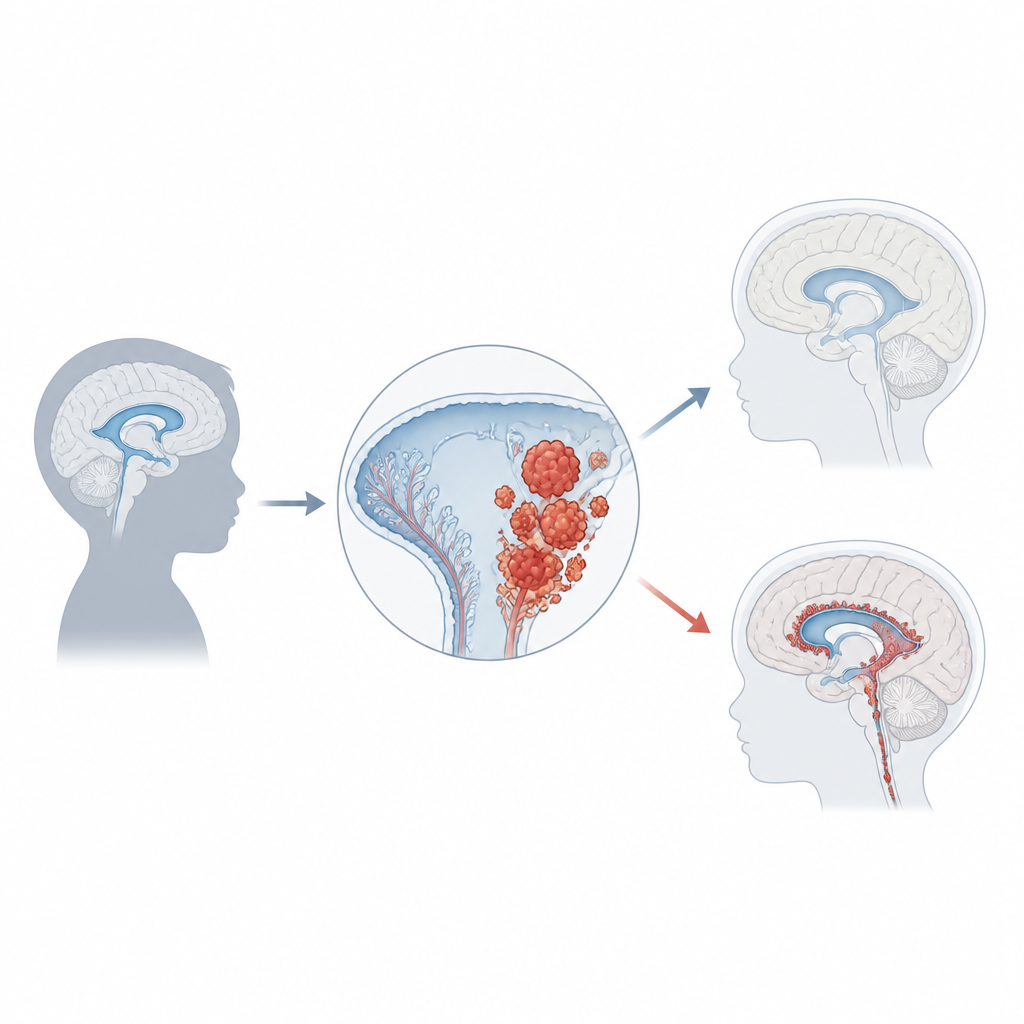
Gènes, régulateurs et risque tumoral
Un des indices les plus forts sur la formation de ce cancer vient de la génétique. Environ la moitié des tumeurs présentent des altérations de TP53, un gène qui stoppe normalement la croissance ou déclenche la mort cellulaire quand l’ADN est endommagé. Les enfants qui héritent de mutations TP53 via le syndrome de Li‑Fraumeni sont particulièrement à risque, et leurs tumeurs présentent souvent de nombreux autres changements de l’ADN. Ces patients ont tendance à un pronostic plus défavorable que ceux dont TP53 est intact. D’autres gènes et voies favorisant la prolifération ou la résistance à la mort, notamment MYC, Notch et Wnt, sont également fréquemment perturbés. Outre les mutations d’ADN, des marques chimiques sur l’ADN qui régulent l’activité des gènes forment des profils de « méthylation » distincts, et un profil courant dans ces tumeurs se corrèle avec un comportement plus agressif. Ensemble, ces empreintes génétiques et épigénétiques commencent à classer les patients en sous‑groupes biologiquement significatifs.
Comment les médecins le traitent aujourd’hui
À ce jour, le facteur unique le plus important associé à la survie est l’étendue de l’exérèse que les chirurgiens peuvent réaliser en toute sécurité. Les enfants dont les tumeurs sont complètement ou quasi complètement réséquées s’en sortent bien mieux que ceux qui gardent d’importants résidus. Parce que le cancer se propage souvent via les voies liquidiennes du cerveau, de nombreuses équipes ajoutent de la chimiothérapie et parfois de la radiothérapie après la chirurgie. Pourtant, la radiothérapie peut gravement nuire au cerveau en développement, et elle est particulièrement risquée chez les enfants porteurs de mutations TP53, prédisposés aux seconds cancers induits par les radiations. En conséquence, les cliniciens testent des combinaisons de chimiothérapies intensives visant à contrôler la tumeur tout en épargnant ou en retardant la radiothérapie, surtout chez les plus jeunes patients. Les résultats précoces suggèrent que certains cocktails médicamenteux peuvent aider, mais le meilleur protocole dépend probablement du profil moléculaire propre à chaque tumeur.
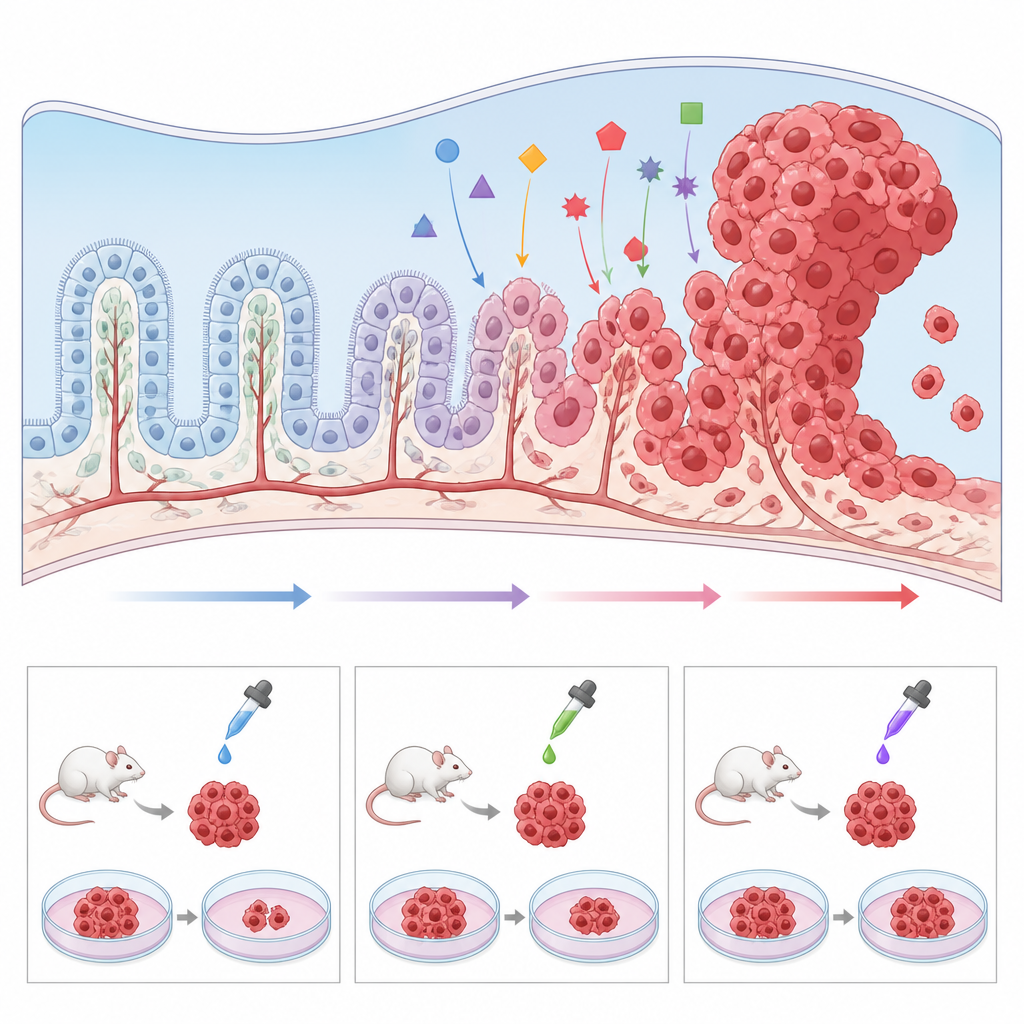
Construire de meilleurs modèles en laboratoire
Parce que le carcinome du plexus choroïde est si rare, les hôpitaux individuels voient très peu de cas et les échantillons tumoraux sont rares. Pour contourner cet obstacle, les chercheurs développent une série de modèles précliniques. Des souris génétiquement modifiées portant les mêmes mutations que celles observées chez l’enfant, telles que la perte de TP53 et l’activation de MYC, développent des tumeurs dans les mêmes régions cérébrales et permettent d’étudier en détail comment des cellules normales du plexus choroïde sont poussées vers le cancer. Les tissus tumoraux peuvent aussi être transplantés chez la souris ou le poisson zèbre pour tester des médicaments dans un système vivant. Parallèlement, les scientifiques ont cultivé des lignées cellulaires tumorales humaines en culture, ainsi que des « mini‑plexus choroïdes » dérivés de cellules souches, et ont montré que la modulation de voies comme Wnt peut transformer un tissu de type sain en poussée de type cancéreux. Ces modèles rendent possible le criblage à haut débit de candidats médicaments et l’exploration des raisons pour lesquelles certaines tumeurs résistent aux thérapies standards.
Vers des traitements plus personnalisés
L’article conclut que de réels progrès contre ce cancer infantile viendront de l’association d’un profilage génétique et épigénétique soigneux de la tumeur de chaque patient à des modèles de laboratoire puissants qui reflètent ces caractéristiques. En identifiant quelles mutations, quels reconfigurations des circuits et quels types cellulaires pilotent chaque cas, les chercheurs espèrent assortir les enfants à des combinaisons médicamenteuses mieux adaptées et concevoir des essais évitant l’usage systématique de radiations sévères. Bien que le carcinome du plexus choroïde restera probablement une maladie rare, ces outils émergents tracent une voie vers une survie plus longue et une meilleure qualité de vie pour les enfants touchés.
Citation: Thompson, A., Pescaru, H., Griffin, B. et al. Choroid plexus carcinoma: state of the field and emerging directions. Oncogenesis 15, 21 (2026). https://doi.org/10.1038/s41389-026-00612-6
Mots-clés: carcinome du plexus choroïde, tumeur cérébrale pédiatrique, mutation TP53, modèles du cancer cérébral, oncologie de précision