Clear Sky Science · de
Karzinom des Plexus choroideus: Stand der Forschung und neue Richtungen
Warum dieser seltene kindliche Hirnkrebs wichtig ist
Das Karzinom des Plexus choroideus ist ein seltener, aber hochgradig aggressiver Hirntumor, der meist sehr junge Kinder trifft, oft noch vor dem Kindergartenalter. Da weltweit nur wenige Patienten betroffen sind, gibt es nur begrenzte Daten, die Ärztinnen und Ärzten Orientierung geben, und die Standardbehandlungen können Überlebende mit lebenslangen Nebenwirkungen belasten. Diese Übersichtsarbeit fasst zusammen, was Forschende und Kliniker heute über die Erkrankung wissen, wie sie entsteht, wie sie derzeit behandelt wird und wie neue Labor‑Modelle möglicherweise endlich den Weg zu sichereren, gezielteren Therapien eröffnen.
Die Flüssigkeitsfabrik des Gehirns und wie sie fehlgeht
Tief in den mit Flüssigkeit gefüllten Räumen des Gehirns liegt ein dünnes, gefranstes Gewebe, der Plexus choroideus. Seine Hauptaufgabe ist die Produktion von Liquor cerebrospinalis, der das Gehirn polstert und Abfallstoffe abtransportiert. Das Gewebe besteht aus spezialisierten Epithelzellen, die kleine Blutgefäße umschließen und durch enge Barrieren vom Liquor getrennt sind. Anders als die meisten Nervenzellen können sich Zellen des Plexus choroideus langsam ein Leben lang teilen, was dem Gewebe erlaubt, sich nach Verletzungen zu reparieren. Diese Fähigkeit zur Teilung macht es jedoch auch anfällig dafür, bei Schädigung zentraler Kontrollgene krebsartig zu werden.
Ein seltener Tumor mit großer Auswirkung auf Kinder
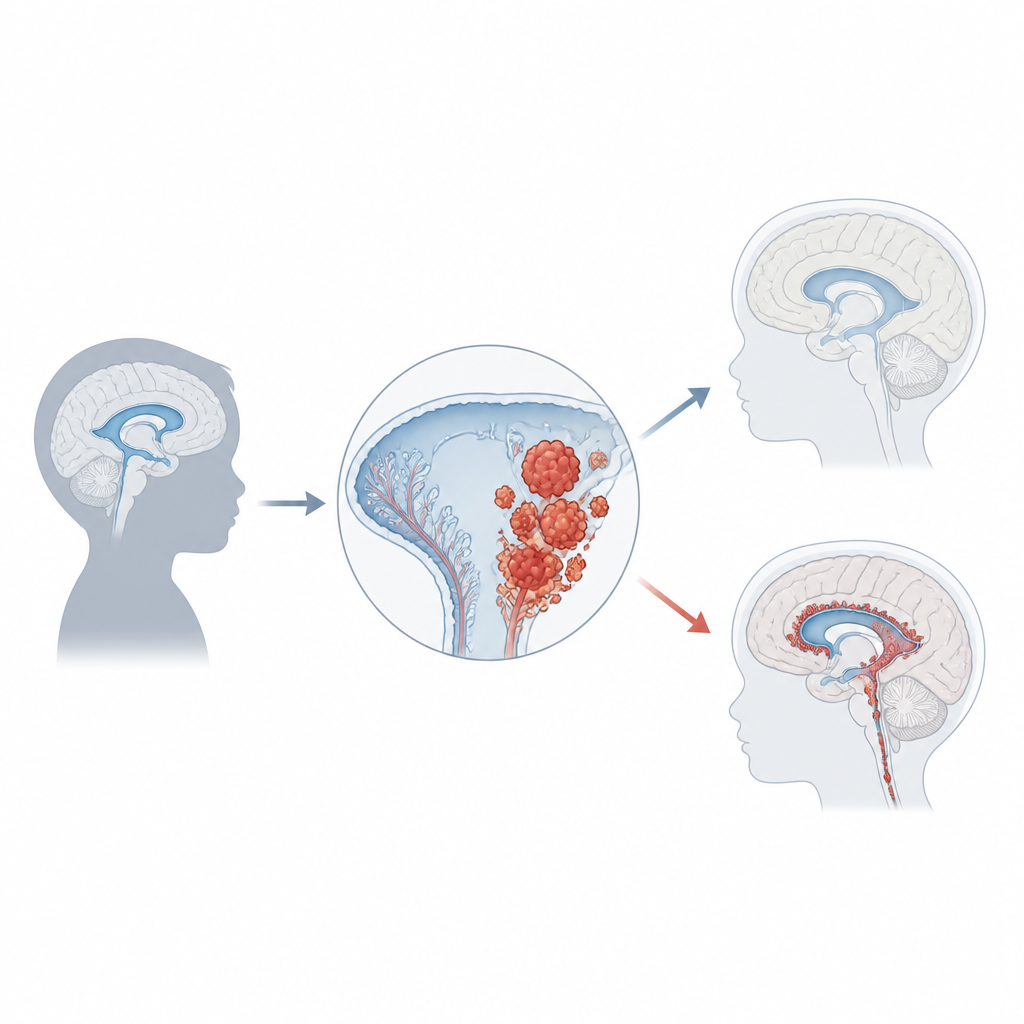
Das Karzinom des Plexus choroideus macht nur etwa 1 Prozent der kindlichen Hirntumoren aus, bei Säuglingen unter einem Jahr kann es jedoch ein Fünftel aller Fälle ausmachen. Die meisten Kinder werden etwa im Alter von drei Jahren diagnostiziert; die Tumoren entstehen meist in den seitlichen oder hinteren Liquorräumen des Gehirns. Beim Wachsen blockieren sie häufig den normalen Fluss der Flüssigkeit und führen so zu einem Druckaufbau, der Hydrozephalus genannt wird. Familien und Ärztinnen und Ärzte bemerken oft zunächst schnelles Kopfwachstum, Erbrechen, Sehprobleme, Kopfschmerzen, Krampfanfälle oder Verhaltensänderungen. Bildgebende Verfahren zeigen große, unregelmäßige Massen, die vom Plexus choroideus ausgehen; eine gesicherte Diagnose erfordert jedoch die mikroskopische Untersuchung und moderne molekulare Tests.
Gene, Schalter und Tumorrisiko
Ein starker Hinweis auf die Entstehung dieses Krebses kommt aus der Genetik. Ungefähr die Hälfte der Tumoren weist Schäden im TP53‑Gen auf, einem Gen, das normalerweise das Zellwachstum stoppt oder Zelltod auslöst, wenn DNA geschädigt ist. Kinder, die TP53‑Mutationen im Rahmen des Li‑Fraumeni‑Syndroms vererbt bekommen, haben ein besonders hohes Risiko, und ihre Tumoren tragen oft viele weitere DNA‑Veränderungen. Diese Patientinnen und Patienten haben tendenziell eine schlechtere Prognose als solche mit intaktem TP53. Weitere Gene und Signalwege, die Zellen zum Wachsen anregen oder sie vor dem Absterben schützen — darunter MYC, Notch und Wnt — sind häufig ebenfalls gestört. Zusätzlich zu DNA‑Mutationen zeigen chemische Markierungen an der DNA, die die Genaktivität regulieren (Methylierung), charakteristische Muster; eines dieser Muster korreliert mit aggressiverem Verhalten. Zusammen beginnen diese genetischen und epigenetischen Fingerabdrücke, Patientengruppen mit biologischer Relevanz zu unterscheiden.
Wie man es heute behandelt
Derzeit ist der wichtigste Faktor, der mit Überleben verknüpft ist, das Ausmaß der Tumorentfernung durch die Chirurgen. Kinder, bei denen der Tumor komplett oder nahezu vollständig entfernt werden kann, schneiden deutlich besser ab als solche mit großen Resten. Da sich der Krebs häufig über die Liquorräume ausbreitet, ergänzen viele Behandlungsteams die Operation durch Chemotherapie und manchmal Bestrahlung. Bestrahlung kann jedoch das sich entwickelnde Gehirn stark schädigen und ist besonders riskant bei Kindern mit TP53‑Mutationen, die anfällig für strahleninduzierte Zweitkrebserkrankungen sind. Deshalb prüfen Kliniker Kombinationen intensiver Chemotherapien, die den Tumor kontrollieren sollen, während Bestrahlung vermieden oder aufgeschoben wird — insbesondere bei den jüngsten Patientinnen und Patienten. Erste Ergebnisse deuten darauf hin, dass bestimmte Wirkstoffkombinationen helfen können, das optimale Regime dürfte jedoch vom molekularen Profil des jeweiligen Tumors abhängen.
Bessere Modelle im Labor aufbauen
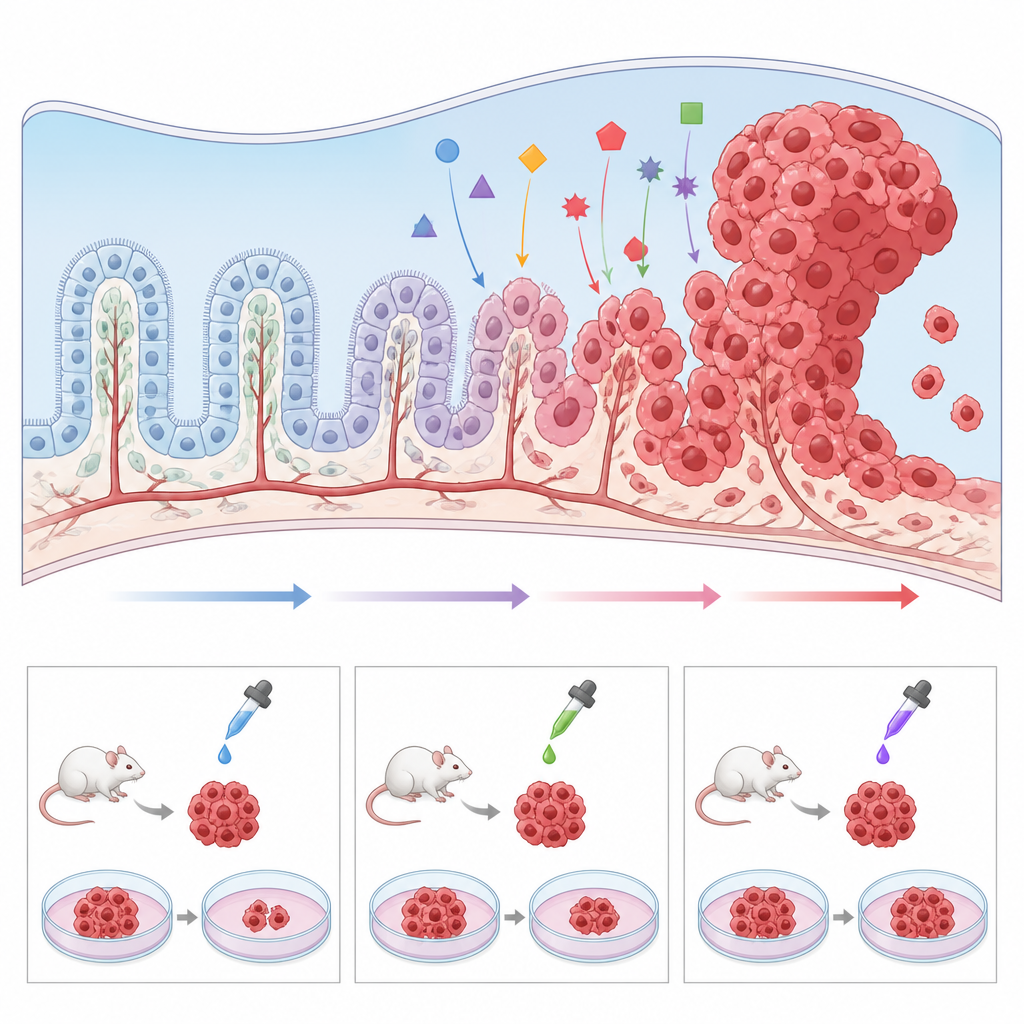
Weil das Karzinom des Plexus choroideus so selten ist, sehen einzelne Krankenhäuser nur wenige Fälle, und Tumormaterial ist knapp. Um dieses Hindernis zu umgehen, entwickeln Forschende eine Reihe präklinischer Modelle. Genetisch veränderte Mäuse, die die gleichen Mutationen wie bei Kindern tragen — etwa TP53‑Verlust und MYC‑Aktivierung — entwickeln Tumoren in denselben Hirnregionen und erlauben eine genaue Untersuchung, wie normale Plexus‑Zellen in Richtung Krebs gedrängt werden. Tumorgewebe kann auch in Mäuse oder Zebrafische transplantiert werden, um Arzneimittel in einem lebenden System zu testen. Parallel dazu haben Wissenschaftler menschliche Tumorzelllinien in Kultur gezogen sowie „Mini‑Plexus choroideus“ aus Stammzellen erzeugt und gezeigt, dass das Verstellen von Signalwegen wie Wnt gesund‑ähnliches Gewebe in tumorförmiges Wachstum verwandeln kann. Diese Modelle ermöglichen Hochdurchsatz‑Screens potenzieller Wirkstoffe und die Erforschung der Ursachen, warum manche Tumoren Standardtherapien widerstehen.
Blick nach vorn: individuellere Behandlung
Der Artikel schließt mit der Einschätzung, dass wirklicher Fortschritt gegen diesen kindlichen Tumor daraus entstehen wird, die sorgfältige genetische und epigenetische Analyse jedes Tumors mit leistungsfähigen Labor‑Modellen zu verknüpfen, die diese Eigenschaften abbilden. Indem man lernt, welche Mutationen, Verschaltungen und Zelltypen jeden Fall antreiben, hoffen Forschende, Kinder mit maßgeschneiderten Wirkstoffkombinationen zu behandeln und Studien zu entwerfen, die den pauschalen Einsatz belastender Bestrahlung vermeiden. Auch wenn das Karzinom des Plexus choroideus wahrscheinlich selten bleiben wird, schaffen diese neuen Werkzeuge einen Weg zu längerem Überleben und besserer Lebensqualität für betroffene Kinder.
Zitation: Thompson, A., Pescaru, H., Griffin, B. et al. Choroid plexus carcinoma: state of the field and emerging directions. Oncogenesis 15, 21 (2026). https://doi.org/10.1038/s41389-026-00612-6
Schlüsselwörter: Karzinom des Plexus choroideus, pädiatrischer Hirntumor, TP53‑Mutation, Modelle für Hirnkrebs, präzisionsonkologie