Clear Sky Science · pl
Ekspansja powtórzeń w genie GOLGA8A jest głównym czynnikiem ryzyka nietypowej czołowo-skroniowej degeneracji płata z inkluzjami pozytywnymi w teobilitynie (aFTLD-U)
Dlaczego to odkrycie dotyczące wczesnego otępienia ma znaczenie
Niewielka, ale wyniszczająca postać otępienia o wczesnym początku dotyka ludzi w najlepszym okresie życia, często w wieku 30., 40. lub 50. lat, zmieniając osobowość i zachowanie znacznie wcześniej niż pojawiają się problemy z pamięcią. Do tej pory lekarze nie wiedzieli, dlaczego większość tych przypadków wydawała się pojawiać nagle, bez wyraźnej historii rodzinnej czy oczywistego genu sprawczego. Badanie to ujawnia główny czynnik genetycznego ryzyka dla jednej z takich postaci — zwanej nietypową czołowo-skroniową degeneracją płata z inkluzjami pozytywnymi w teobilitynie (aFTLD-U) — wskazując na osobliwy odcinek DNA, który u wielu pacjentów jest patologicznie zreplikowany. Praca ta otwiera nowe okno na to, jak ukryte niestabilności w naszym genomie mogą po cichu przygotowywać grunt pod ciężkie choroby mózgu.
Rzadkie otępienie uderzające w przednie części mózgu
Otępienie czołowo-skroniowe (FTD) jest jedną z najczęstszych przyczyn otępienia przed 65. rokiem życia. W przeciwieństwie do choroby Alzheimera, która głównie dotyka pamięć, FTD często zaczyna się od dramatycznych zmian w osobowości, osądzaniu i zachowaniu społecznym. aFTLD-U to rzadka patologiczna podgrupa w obrębie tego spektrum. Osoby z aFTLD-U zazwyczaj rozwijają ciężkie objawy behawioralne, przy relatywnie zachowanej mowie i ruchu. Badania obrazowe mózgu wykazują wyraźne zaniki płatów czołowych i struktur głębokich mózgu, które pomagają kontrolować motywację i nawyki. Pod mikroskopem komórki mózgowe gromadzą nieprawidłowe skupiska niektórych białek wiążących RNA, znanych łącznie jako białka FET. Jednak w przeciwieństwie do innych podtypów FTD, nie zidentyfikowano dotąd jednoznacznego genu sprawczego u tych pacjentów, którzy w większości wydawali się być przypadkami «sporadycznymi».
W poszukiwaniu ukrytego regionu ryzyka w genomie
Aby odnaleźć genetyczne składniki choroby, naukowcy stworzyli międzynarodowy konsorcjum i zgromadzili DNA oraz próbki mózgów od osób, u których aFTLD-U zostało jednoznacznie zdiagnozowane podczas autopsji. Porównali 59 takich przypadków z ponad 3000 osobami kontrolnymi, stosując badanie asocjacji genomowej (GWAS), przesiew poszukujący różnic w milionach markerów DNA między pacjentami a kontrolami. Jeden region na chromosomie 15 (oznaczony jako 15q14) dał wyjątkowo silny sygnał, wskazując na dwa powiązane wzorce zmienności genetycznej, czyli haplotypy, które występowały znacznie częściej u pacjentów niż u zdrowych osób. Haplotypy ryzyka leżą blisko pary niemal identycznych genów, GOLGA8A i GOLGA8B, w części genomu, która jest notorycznie trudna do analizy z powodu wielu zduplikowanych i przearanżowanych segmentów.
Długi, niestabilny powtórzeniowy odcinek DNA okazuje się kluczowy
Ponieważ standardowe sekwencjonowanie krótkich odczytów ma trudności w tej złożonej części genomu, zespół sięgnął po sekwencjonowanie długich odczytów, które pozwala odczytać znacznie większe fragmenty DNA za jednym razem. Analizując ponad 1700 genomów, w tym wiele przypadków aFTLD-U oraz grup porównawczych z innymi chorobami mózgu, odkryli, że haplotypy ryzyka niemal zawsze niosły ze sobą wyjątkowo długi «tandemowy powtórzenie» wewnątrz intronu genu GOLGA8A. Powtórzenia tandemowe przypominają zacięcia w sekwencji DNA, gdzie małe jednostki liter są kopiowane wielokrotnie. U zdrowych osób ten powtórzenie jest krótkie; u wielu pacjentów z aFTLD-U było ono masywnie rozszerzone, czasem obejmując ponad 2000 par zasad. Te rozszerzone wersje nie były identyczne, ale te najsilniej powiązane z chorobą dominowały przez prosty wzorzec dwu-literowy oparty na nukleotydach C i T powtarzanych w parach. Gdy badacze zaklasyfikowali nosicieli według długości powtórzenia i procentowej zawartości CT, długie, bogate w CT ekspansje przewidywały aFTLD-U znacznie lepiej niż pierwotne warianty markerowe.
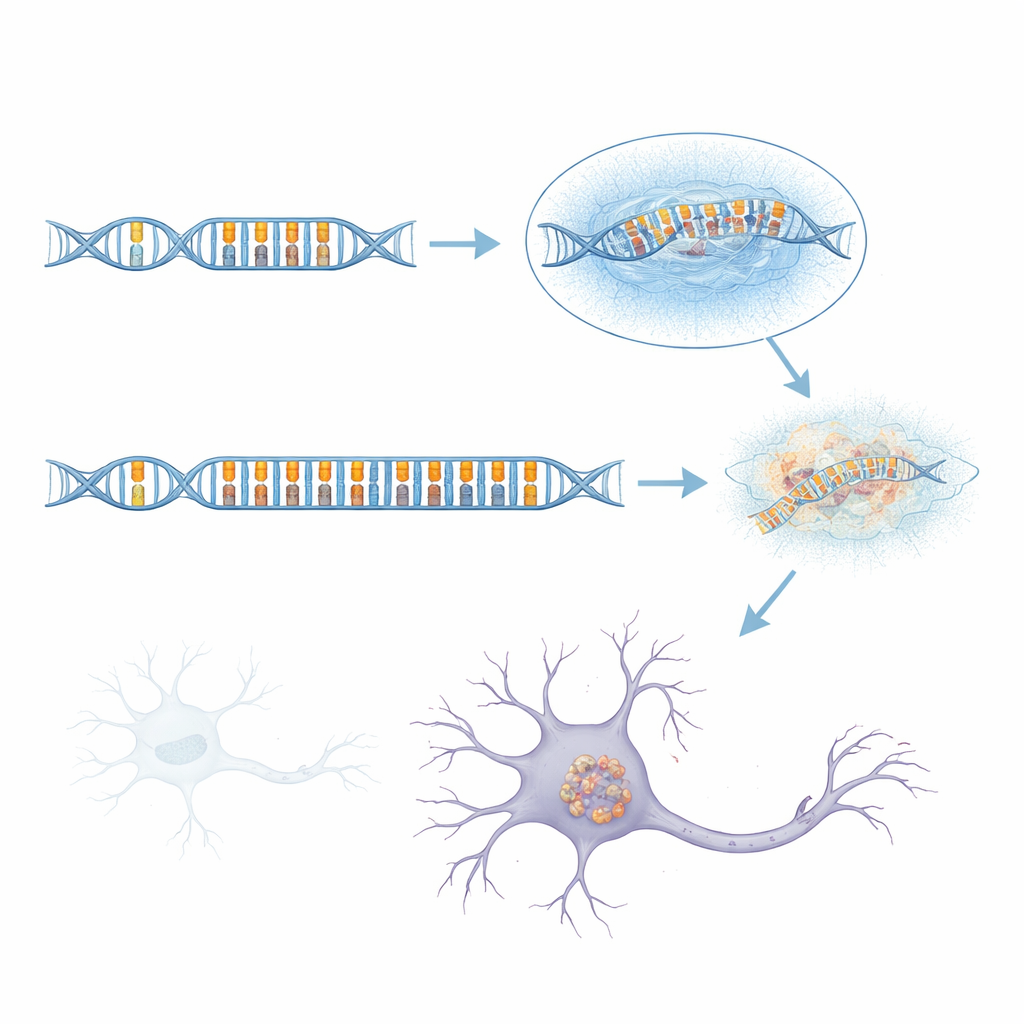
Zmienność, dziedziczenie i subtelne ryzyko
Historia nie jest tak prosta, jak pojedyncza mutacja, która zawsze powoduje chorobę. Niektórzy ludzie z populacji ogólnej, w tym krewni pacjentów z aFTLD-U, nosili duże, bogate w CT ekspansje w genie GOLGA8A, a mimo to nie rozwinęli otępienia — przynajmniej w momencie badania. Rozszerzone powtórzenie wykazywało też oznaki niestabilności: jego długość różniła się między komórkami i między tkankami, co sugeruje, że może ono dalej rosnąć lub kurczyć się w trakcie życia osoby. Co ciekawe, około 70% pacjentów z aFTLD-U w kohorcie stanowili mężczyźni, co sugeruje, że czynniki biologiczne związane z płcią lub narażenia środowiskowe mogą wpływać na to, czy nosiciel faktycznie zachoruje. Ponadto około 40% potwierdzonych przypadków aFTLD-U nie wykazywało w ogóle tej konkretnej ekspansji, co implikuje, że inne, jeszcze nieodkryte czynniki genetyczne lub środowiskowe mogą doprowadzić do tej samej patologii mózgowej.
Co to oznacza dla zrozumienia i diagnozowania choroby
Chociaż naukowcy jeszcze nie wiedzą, jak to intronowe powtórzenie GOLGA8A szkodzi komórkom mózgu, jego lokalizacja i skład sugerują kilka możliwości. Rozszerzony fragment DNA może zmieniać sposób, w jaki pobliskie geny są włączane i wyłączane, produkować nietypowe cząsteczki RNA, które wiążą ważne białka, lub tworzyć toksyczne struktury wewnątrz neuronów. Jaki by nie był dokładny mechanizm, obecność bardzo długiego, bogatego w CT powtórzenia w niemal 60% przypadków aFTLD-U czyni z niego główny czynnik ryzyka, porównywalny z silnymi wpływami genetycznymi w bardziej znanych chorobach neurodegeneracyjnych. W praktyce testowanie haplotypów na chromosomie 15 i powtórzenia w GOLGA8A może pomóc zidentyfikować osoby z wczesnymi objawami behawioralnymi, które prawdopodobnie mają ten konkretny podtyp FTD, wspierając diagnozę i badania. Szerzej, ta praca podkreśla, jak ukryte ekspansje powtórzeń — długie odcinki powtarzanego DNA ukryte w trudnym terenie genomowym — mogą leżeć u podstaw pozornie sporadycznych zaburzeń mózgowych i toruje drogę do przyszłych badań mających na celu rozwikłanie ich biologii, a potencjalnie też ukierunkowanie terapii.
Cytowanie: De Coster, W., Van den Broeck, M., Baker, M. et al. A repeat expansion in GOLGA8A is a major risk factor for atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions. Nat Genet 58, 726–736 (2026). https://doi.org/10.1038/s41588-026-02537-7
Słowa kluczowe: otępienie czołowo-skroniowe, ekspansja powtórzeń, GOLGA8A, neurodegeneracja, ryzyko genetyczne