Clear Sky Science · it
Una espansione di ripetizioni in GOLGA8A è un importante fattore di rischio per la degenerazione lobare frontotemporale atipica con inclusioni ubichitinate
Perché questa scoperta sulla demenza precoce è importante
Una forma rara ma devastante di demenza a esordio precoce colpisce persone nel pieno della vita, spesso tra i 30 e i 50 anni, alterando personalità e comportamento molto prima che compaiano problemi di memoria. Finora i medici non sapevano perché la maggior parte di questi casi sembrasse insorgere dal nulla, senza una chiara storia familiare o un gene responsabile. Questo studio mette in luce un importante fattore di rischio genetico per una di queste condizioni — chiamata degenerazione lobare frontotemporale atipica con inclusioni ubichitinate (aFTLD-U) — individuando un tratto di DNA anomalo ripetuto in molti pazienti. Il lavoro apre una nuova finestra su come instabilità nascoste nel nostro genoma possano silenziosamente predisporre a gravi malattie cerebrali.
Una demenza rara che colpisce la porzione frontale del cervello
La demenza frontotemporale (FTD) è una delle cause più comuni di demenza prima dei 65 anni. Diversamente dalla malattia di Alzheimer, che interessa principalmente la memoria, la FTD spesso inizia con cambiamenti drammatici nella personalità, nel giudizio e nel comportamento sociale. L’aFTLD-U è un sottotipo patologico raro all’interno di questo gruppo. Le persone con aFTLD-U di solito sviluppano sintomi comportamentali severi ma conservano relativamente il linguaggio e il movimento. Le immagini cerebrali mostrano un evidente restringimento dei lobi frontali e di strutture profonde che aiutano a controllare motivazione e abitudini. Al microscopio, le cellule cerebrali colpite accumulano aggregati anormali di certe proteine leganti l’RNA, note collettivamente come proteine FET. Tuttavia, a differenza di altri sottotipi di FTD, non era stato identificato un gene-causing chiaro per questi pazienti, che per lo più apparivano come casi “sporadici”.
Alla ricerca di una regione di rischio nascosta nel genoma
Per individuare contributi genetici, i ricercatori hanno formato un consorzio internazionale e raccolto campioni di DNA e tessuto cerebrale da persone diagnosticate in modo definitivo con aFTLD-U all’autopsia. Hanno confrontato 59 casi di questo tipo con più di 3.000 individui di controllo tramite uno studio di associazione su scala genomica, una scansione che cerca milioni di marcatori del DNA per differenze tra pazienti e controlli. Una regione sul cromosoma 15 (chiamata 15q14) ha mostrato un segnale eccezionalmente forte, indicando due pattern correlati di variazione genetica, o aplotipi, molto più comuni nei pazienti che nelle persone sane. Questi aplotipi di rischio si trovano vicino a una coppia di geni quasi identici, GOLGA8A e GOLGA8B, in una parte del genoma notoriamente difficile da analizzare perché contiene molti segmenti duplicati e riorganizzati.
Una lunga ripetizione di DNA instabile emerge come fattore chiave
Poiché il sequenziamento a letture corte standard fatica in questa regione complessa, il team si è rivolto al sequenziamento a letture lunghe, che può leggere porzioni di DNA molto più grandi in un’unica passata. Esaminando più di 1.700 genomi, inclusi molti casi di aFTLD-U e gruppi di confronto con altre malattie cerebrali, hanno scoperto che gli aplotipi di rischio quasi sempre portavano una “ripetizione tandem” insolitamente lunga all’interno di un introne del gene GOLGA8A. Le ripetizioni tandem somigliano a balbuzie nella sequenza del DNA, dove piccole unità di lettere vengono copiate più volte. Nelle persone sane questa ripetizione è corta; in molti pazienti con aFTLD-U era enormemente espansa, talvolta estendendosi per oltre 2.000 basi di DNA. Queste versioni espanse non erano tutte identiche, ma quelle più fortemente associate alla malattia erano dominate da un semplice motivo di due lettere basato sui nucleotidi C e T ripetuti a coppie. Quando i ricercatori classificarono i portatori in base alla lunghezza della ripetizione e alla percentuale di contenuto CT, le espansioni lunghe e ricche di CT predicevano l’aFTLD-U molto meglio dei marcatori originali.
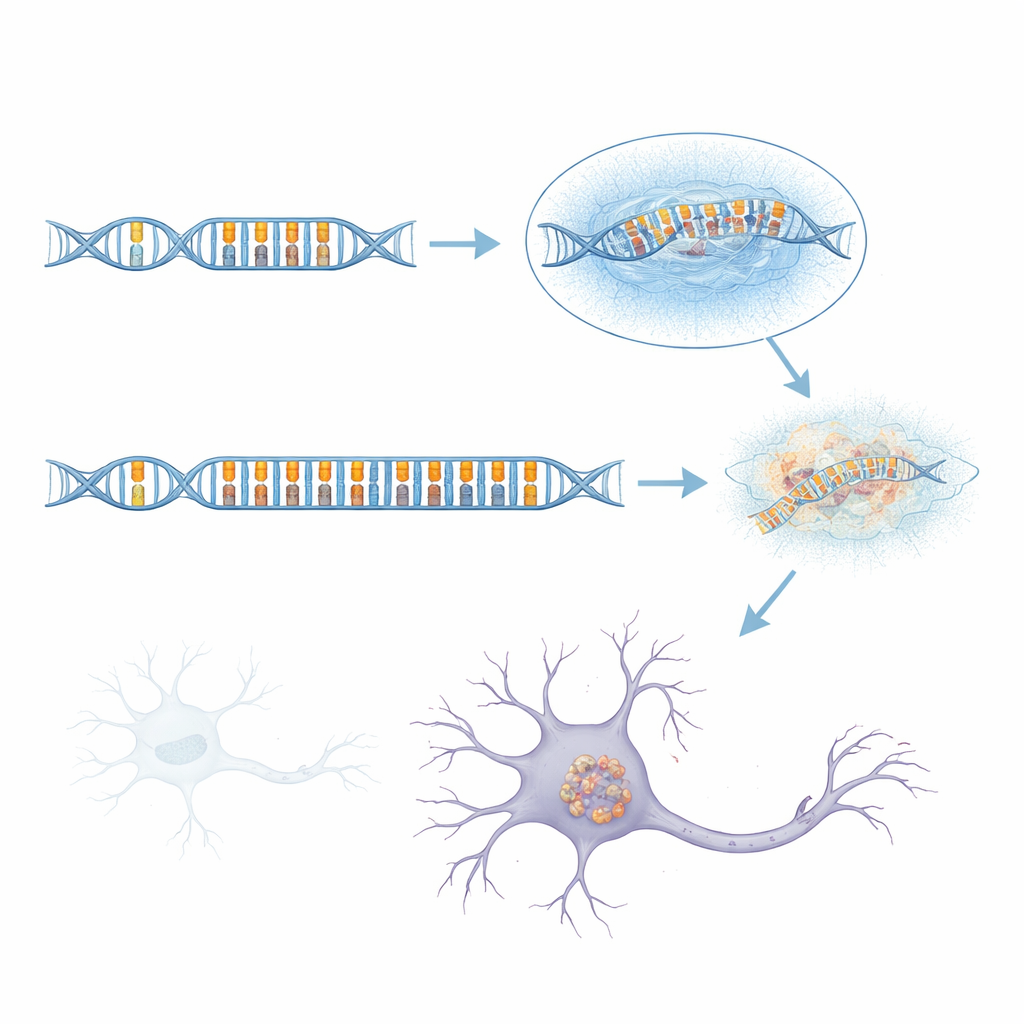
Variabilità, ereditarietà e rischio sottile
La storia non è semplice come una singola mutazione che causa sempre la malattia. Alcune persone nella popolazione generale, compresi parenti di pazienti con aFTLD-U, portavano grandi espansioni ricche di CT in GOLGA8A e tuttavia non avevano sviluppato demenza — almeno al momento dello studio. La ripetizione espansa mostrava anche segni di instabilità: la sua lunghezza variava da cellula a cellula e da tessuto a tessuto, suggerendo che può continuare a crescere o restringersi nel corso della vita di una persona. È interessante che circa il 70% dei pazienti con aFTLD-U nella coorte fosse di sesso maschile, suggerendo che fattori biologici legati al sesso o esposizioni ambientali possano influenzare se un portatore sviluppi o meno la malattia. Inoltre, approssimativamente il 40% dei casi confermati di aFTLD-U non mostrava affatto questa espansione particolare, implicando che altri fattori genetici o ambientali, ancora da scoprire, possano produrre la stessa patologia cerebrale.
Cosa significa per la comprensione e la diagnosi della malattia
Pur non sapendo ancora come questa ripetizione intronica di GOLGA8A danneggi le cellule cerebrali, la sua posizione e composizione suggeriscono diverse possibilità. Il DNA espanso potrebbe alterare il modo in cui i geni vicini vengono accesi e spenti, produrre molecole di RNA insolite che intrappolano proteine vitali o formare strutture tossiche all’interno dei neuroni. Qualunque sia il meccanismo preciso, la presenza di una ripetizione molto lunga e ricca di CT in quasi il 60% dei casi di aFTLD-U la qualifica come un importante fattore di rischio, alla pari con influenze genetiche forti in malattie neurodegenerative più familiari. In termini pratici, testare gli aplotipi sul cromosoma 15 e la ripetizione in GOLGA8A potrebbe aiutare a identificare persone con sintomi comportamentali precoci che probabilmente hanno questo sottotipo specifico di FTD, facilitando la diagnosi e la ricerca. Più in generale, questo lavoro evidenzia come espansioni criptiche di ripetizioni — lunghe porzioni di DNA ripetuto nascoste in regioni genomiche difficili — possano essere alla base di disturbi cerebrali apparentemente sporadici, e apre la strada a studi futuri per svelarne la biologia e, potenzialmente, rivolgersi a esse terapeuticamente.
Citazione: De Coster, W., Van den Broeck, M., Baker, M. et al. A repeat expansion in GOLGA8A is a major risk factor for atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions. Nat Genet 58, 726–736 (2026). https://doi.org/10.1038/s41588-026-02537-7
Parole chiave: demenza frontotemporale, espansione di ripetizioni, GOLGA8A, neurodegenerazione, rischio genetico