Clear Sky Science · pl
Toksyczność ludzkiego FUS poprzez interakcję z polimerazą RNA II u Drosophila
Dlaczego to ma znaczenie dla zdrowia mózgu
SLA (choroba Lou Gehriga) i niektóre postacie otępienia stopniowo niszczą komórki nerwowe, a wciąż nie rozumiemy w pełni, dlaczego te komórki umierają. W badaniu wykorzystano muszki owocowe, by zbadać białko o nazwie FUS, powiązane z niektórymi dziedzicznymi postaciami SLA i występujące w skupiskach w odmianie otępienia zwanej czołowo-skroniową degeneracją płatów (FTLD). Pytając, gdzie w komórce FUS wyrządza najwięcej szkody i z jakimi partnerami wchodzi w interakcje, badacze odkrywają zaskakującą, jądrową drogę toksyczności, która może pomóc wyjaśnić, dlaczego neurony zawodzą w określonych chorobach mózgu.
Od nerwów muszki do chorób ludzkich
FUS to białko, które normalnie występuje w jądrze komórkowym, gdzie przechowywane jest DNA i odczytywane geny. U pacjentów z powiązaną z FUS postacią SLA, mutant FUS często przemieszcza się do cytoplazmy, tworząc widoczne skupiska. Popularna hipoteza głosiła, że to właśnie cytoplazmatyczne depozyty zabijają neurony. U muszek owocowych jednak samo nadmierne wytwarzanie normalnego ludzkiego FUS w komórkach nerwowych wystarczy, by zaburzyć prawidłowy rozwój, skrócić życie i upośledzić ruch. Sugeruje to, że nadmiar FUS, nawet bez mutacji, jest niebezpieczny i stanowi kontrolowany system do rozdzielenia, gdzie i jak pojawia się ta toksyczność.
Zawrotka w opowieści o przemieszczeniu białka
Zespół skonstruował muszki produkujące wersję FUS pozbawioną wbudowanego „kodu pocztowego” jądrowego, znanego jako sekwencja lokalizacji jądrowej. Bez tej cechy FUS w dużej mierze nie trafia do jądra i zamiast tego gromadzi się w cytoplazmie. Wbrew oczekiwaniom te muszki były mniej chore niż osobniki produkujące normalny FUS: przeżywały dłużej i rozwijały się bardziej prawidłowo, nawet gdy poziomy białka były wyrównane, by obie wersje występowały w podobnych ilościach. Szczegółowe obrazowanie neuronów larwalnych i dorosłych potwierdziło, że standardowy FUS skupiał się głównie w jądrze, podczas gdy zmieniona wersja pozostawała głównie na zewnątrz. Na tej podstawie autorzy wnioskują, że w tym modelu FUS szkodzi neuronom przede wszystkim przez swoją aktywność w jądrze, a nie przez cytoplazmatyczne skupiska.
Ukryte skupiska w centrum kontroli komórki
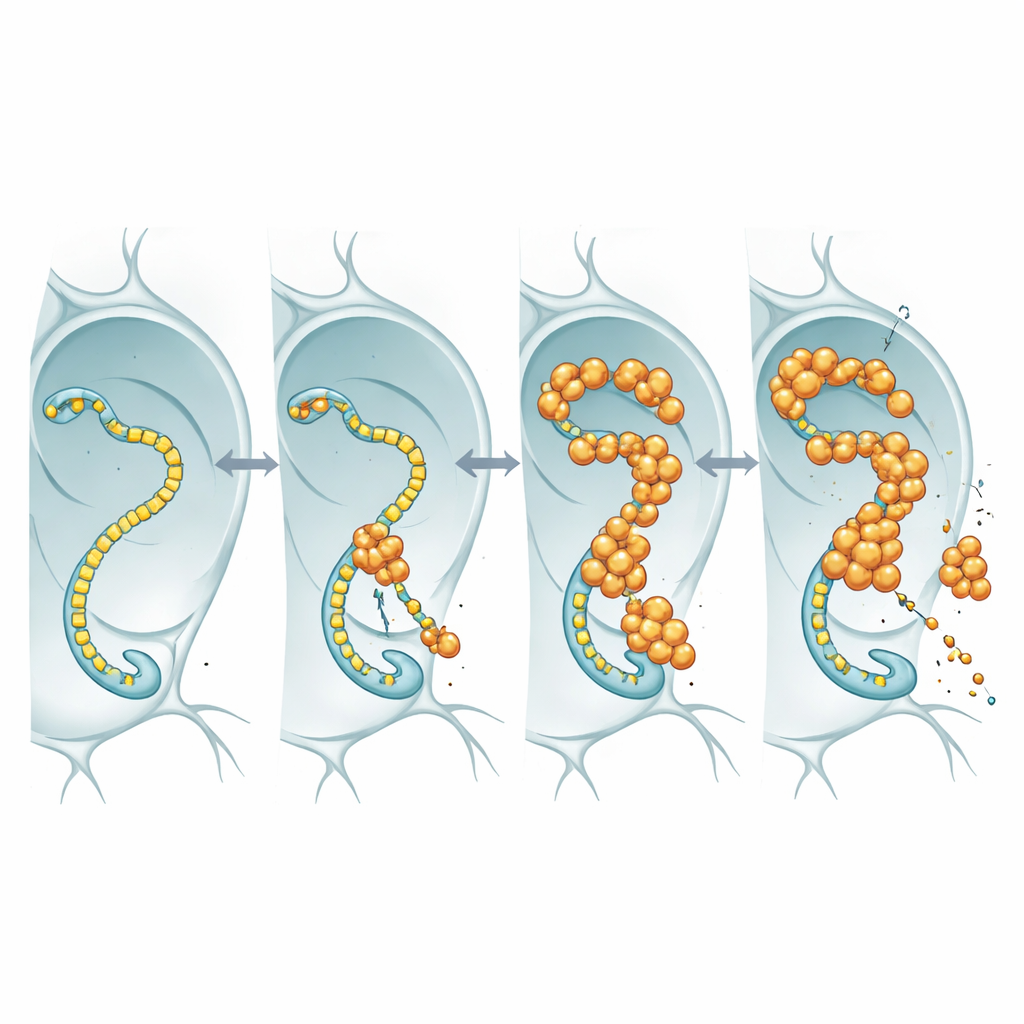
Aby zobaczyć, co jądrowy FUS właściwie robi, badacze znakowali FUS fluorescencyjnym markerem i obserwowali go w powiększonych jądrach gruczołu ślinowego muszek oraz w neuronach. Zamiast tworzyć twarde, nierozpuszczalne agregaty, FUS gromadził się w jasnych, punktowych granulkach, które pozostawały dynamiczne: gdy obszar był wybłyszczany laserem, nie wybłyszczone cząsteczki wpływały z powrotem w ciągu sekund. Te krople pojawiały się często w regionach zawierających wysokie poziomy polimerazy RNA II, enzymu, który przesuwa się wzdłuż DNA, tworząc kopie RNA genów. Taka kolokacja sugeruje, że FUS może zakłócać podstawowy mechanizm transkrypcji, który zaopatruje neurony w potrzebne im wiadomości RNA.
Kiedy maszyna odczytująca geny staje się wspólnikiem w zbrodni
Polimeraza RNA II posiada elastyczny ogon zbudowany z powtarzanych krótkich motywów; ogon ten działa jak pas startowy dla wielu regulatorów, w tym FUS i jego krewniaków. Wykorzystując linie muszek, w których ogon ten został zmodyfikowany tak, by zawierać mniej lub więcej powtórzeń, zespół sprawdził, czy zmiana długości ogona wpłynie na toksyczność FUS. U muszek nadprodukujących FUS skrócenie ogona wydłużyło życie zwierząt, mimo że poziomy RNA i białka FUS pozostawały podobne. Zależność od długości odpowiada wcześniejszym pracom biochemicznym pokazującym, że dłuższe ogony wiążą FUS silniej. Najprostsza interpretacja jest taka, że FUS wywiera toksyczny efekt poprzez angażowanie tego ogona: gdy ogon jest długi, FUS wiąże się mocniej, zaburza normalną kontrolę transkrypcji i neurony cierpią; gdy ogon jest skrócony, FUS ma mniej miejsc do uchwytu i staje się mniej szkodliwy.
Wskazówki z ludzkiej tkanki mózgowej
Autorzy zwrócili się następnie do ludzkich próbek pośmiertnych. W tkance rdzenia kręgowego od pacjentów z SLA niosącymi mutacje FUS zaobserwowali oczekiwane inkluzje pozytywne dla FUS w neuronach, ale ludzka wersja dużej podjednostki tego enzymu (POLR2A) pozostawała w zwykłej, jądrowej lokalizacji. Dla odmiany, w korze czołowej u pacjentów z FTLD oznaczonym inkluzjami pozytywnymi dla FUS, POLR2A często pojawiał się w nietypowych cytoplazmatycznych punktach, które czasem nakładały się na ogniska FUS. Te przemieszczania nie występowały w mózgach kontrolnych. Wzorzec ten sugeruje, że w FTLD, ale niekoniecznie w FUS-SLA, FUS może ściągać polimerazę RNA II poza jej właściwe miejsce, potencjalnie paraliżując ekspresję genów w dotkniętych neuronach.
Co to oznacza dla przyszłych terapii
Podsumowując, praca wskazuje, że w tych modelach muszek najgroźniejszą postacią FUS nie jest cytoplazmatyczne skupisko, lecz wersja jądrowa, która wchodzi w interakcje z maszynerią odczytu genów. Poprzez tworzenie dynamicznych granulek w pobliżu polimerazy RNA II i wiązanie jej elastycznego ogona, nadmiar FUS wydaje się zakłócać przepływ informacji genetycznej potrzebnej do utrzymania neuronów przy życiu. Obserwacja, że POLR2A również ulega przemieszczeniu w tkance mózgowej ludzi z FTLD, wzmacnia argument, że ta jądrowa współpraca przyczynia się do choroby. Terapie ograniczające gromadzenie się FUS w jądrze lub osłabiające jego przyczepność do polimerazy RNA II mogłyby zatem chronić neurony, oferując nowe podejście terapeutyczne wobec chorób związanych z SLA i otępień z obecnością FUS.
Cytowanie: Moens, T.G., Biasetti, L., Scheveneels, W. et al. Human FUS is toxic via association with RNA polymerase II in Drosophila. Cell Death Dis 17, 310 (2026). https://doi.org/10.1038/s41419-026-08539-x
Słowa kluczowe: białko FUS, polimeraza RNA II, SLA, otępienie czołowo-skroniowe, model Drosophila