Clear Sky Science · it
La FUS umana è tossica per associazione con l’RNA polimerasi II in Drosophila
Perché è importante per la salute del cervello
La SLA (malattia di Lou Gehrig) e alcune forme di demenza distruggono progressivamente i neuroni, eppure non comprendiamo ancora pienamente perché queste cellule muoiano. Questo studio utilizza la Drosophila per analizzare una proteina chiamata FUS, collegata ad alcuni casi ereditari di SLA e presente anche in aggregati in una forma di demenza chiamata degenerazione lobare frontotemporale (FTLD). Cercando di capire dove nella cellula la FUS causi il maggior danno e con quali partner interagisca, i ricercatori rivelano una sorprendente via nucleare di tossicità che potrebbe aiutare a spiegare perché i neuroni falliscono in specifiche malattie cerebrali.
Dai nervi della mosca alla malattia umana
La FUS è una proteina che normalmente risiede nel nucleo cellulare, dove è immagazzinato il DNA e dove i geni vengono trascritti. Nei pazienti con SLA legata a FUS, la FUS mutata spesso si sposta nel citoplasma, formando ammassi visibili. Un’idea diffusa è che siano questi depositi citoplasmatici a uccidere i neuroni. Nella Drosophila, tuttavia, la sola sovraespressione della FUS umana normale nei neuroni è già sufficiente a bloccare uno sviluppo corretto, accorciare la vita e danneggiare il movimento. Questo suggerisce che un eccesso di FUS, anche senza mutazione, sia pericoloso e fornisce un sistema controllato per scomporre come e dove emerge questo pericolo.
Un colpo di scena nella storia dello spostamento proteico
Il gruppo ha generato mosche che producono una versione di FUS priva del suo “codice postale” nucleare, noto come sequenza di localizzazione nucleare. Senza questo segnale, la FUS non riesce in gran parte a entrare nel nucleo e si accumula invece nel citoplasma. Contrariamente alle aspettative, queste mosche erano meno malate rispetto alle mosche che esprimevano la FUS normale: sopravvivevano meglio e si sviluppavano in modo più regolare, anche quando i livelli proteici erano regolati in modo che le due versioni fossero presenti in quantità simili. Imaging dettagliato dei neuroni larvali e adulti ha confermato che la FUS standard si accumulava principalmente nel nucleo, mentre la versione alterata rimaneva per lo più all’esterno. Questi confronti hanno portato gli autori a concludere che, in questo modello, la FUS danneggia i neuroni principalmente per ciò che fa nel nucleo, non per gli ammassi citoplasmatici.
Aggregati nascosti nella sala di controllo della cellula
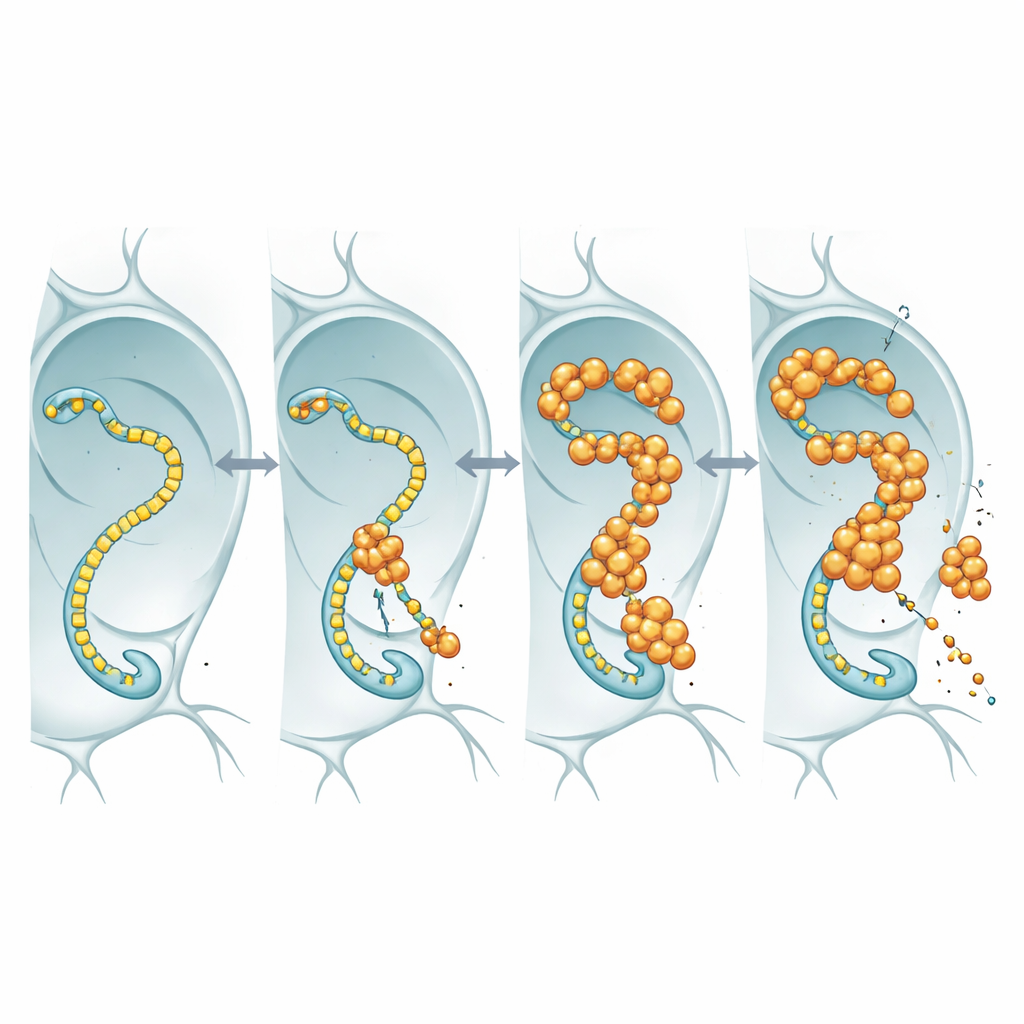
Per capire cosa stia effettivamente facendo la FUS nucleare, i ricercatori hanno marcato la FUS con un marker fluorescente e l’hanno osservata nei nuclei sovradimensionati delle ghiandole salivari delle mosche e nei neuroni. Piuttosto che formare aggregati duri e insolubili, la FUS si raccoglieva in granuli puntiformi e brillanti che restavano dinamici: quando una zona veniva sbiancata con un laser, molecole non sbiancate affluivano di nuovo in pochi secondi. Queste goccioline tendevano a comparire in regioni che contenevano anche alti livelli di RNA polimerasi II, l’enzima che percorre il DNA per produrre copie di RNA dei geni. Questa co-localizzazione ha suggerito che la FUS potrebbe interferire con la macchina trascrizionale di base che fornisce ai neuroni i messaggi di RNA necessari.
Quando la macchina che legge i geni diventa complice
L’RNA polimerasi II possiede una coda flessibile composta da brevi motivi ripetuti; questa coda funge da pista di atterraggio per molti fattori regolatori, compresa la FUS e proteine affini. Usando linee di mosche in cui questa coda era ingegnerizzata per contenere meno o più ripetizioni, il gruppo ha chiesto se variare la lunghezza della coda altersse la tossicità della FUS. Nelle mosche che sovraproducevano FUS, l’accorciamento della coda aumentava la durata di vita degli animali, nonostante i livelli di RNA e proteina FUS rimanessero simili. Questa dipendenza dalla lunghezza corrisponde a lavori biochimici precedenti che mostrano che code più lunghe legano la FUS con maggior affinità. L’interpretazione più semplice è che la FUS eserciti il suo effetto tossico impegnando questa coda: quando la coda è lunga, la FUS si lega più fortemente, altera il controllo trascrizionale normale e i neuroni ne risentono; quando la coda è più corta, la FUS ha meno punto di ancoraggio e diventa meno dannosa.
Indizi dal tessuto cerebrale umano
Gli autori si sono poi rivolti a campioni post-mortem umani. Nel midollo spinale di pazienti con SLA portatori di mutazioni in FUS, hanno osservato le attese inclusioni positive per FUS nei neuroni, ma la sottounità maggiore dell’enzima umano (POLR2A) è rimasta nella sua consueta posizione nucleare. Al contrario, nella corteccia frontale di pazienti con FTLD caratterizzata da inclusioni positive per FUS, POLR2A appariva spesso in punti citoplasmatici anomali che talvolta sovrapponevano i depositi di FUS. Questi spostamenti erano assenti nei cervelli di controllo. Questo schema suggerisce che nella FTLD, ma non necessariamente nella SLA da FUS, la FUS possa trascinare l’RNA polimerasi II fuori posto, compromettendo potenzialmente l’espressione genica nei neuroni colpiti.
Cosa significa per terapie future
Nel complesso, il lavoro sostiene che in questi modelli di mosca la forma di FUS più pericolosa non sono gli ammassi citoplasmatici ma la versione nucleare che interagisce con la macchina che legge i geni. Formando granuli dinamici vicino all’RNA polimerasi II e legando la sua coda flessibile, l’eccesso di FUS sembra disturbare il flusso di informazione genetica necessario per mantenere vivi i neuroni. L’osservazione che POLR2A si mislocalizzi anche nel tessuto cerebrale umano di FTLD rafforza l’ipotesi che questa partnership nucleare contribuisca alla malattia. Terapie che limitino l’accumulo di FUS nel nucleo o ne indeboliscano l’interazione con l’RNA polimerasi II potrebbero quindi proteggere i neuroni, offrendo un nuovo approccio per SLA correlate e demenze positive per FUS.
Citazione: Moens, T.G., Biasetti, L., Scheveneels, W. et al. Human FUS is toxic via association with RNA polymerase II in Drosophila. Cell Death Dis 17, 310 (2026). https://doi.org/10.1038/s41419-026-08539-x
Parole chiave: Proteina FUS, RNA polimerasi II, SLA, demenza frontotemporale, modello Drosophila