Clear Sky Science · de
Humanes FUS ist über Assoziation mit RNA-Polymerase II in Drosophila toxisch
Warum das für die Gehirngesundheit wichtig ist
ALS (Lou-Gehrigs Krankheit) und bestimmte Formen der Demenz zerstören langsam Nervenzellen, doch wir verstehen noch nicht vollständig, warum diese Zellen absterben. Diese Studie nutzt Fruchtfliegen, um ein Protein namens FUS zu untersuchen, das mit einigen erblichen ALS-Fällen verbunden ist und in einer Form der Demenz namens frontotemporale lobäre Degeneration (FTLD) in Klumpen vorkommt. Indem die Forschenden fragen, an welchem Ort in der Zelle FUS am schädlichsten ist und mit welchen Partnern es interagiert, entdecken sie einen überraschenden nukleären Weg zur Toxizität, der helfen könnte zu erklären, warum Neurone bei bestimmten Hirnerkrankungen versagen.
Von Fliegennerven zur menschlichen Krankheit
FUS ist ein Protein, das normalerweise im Zellkern vorkommt, wo die DNA lagert und Gene abgelesen werden. Bei Patienten mit FUS-assoziiertem ALS verlagert sich mutiertes FUS häufig ins Zytoplasma und bildet sichtbare Klumpen. Eine verbreitete Idee war, dass diese zytoplasmatischen Ablagerungen die Neurone töten. In Fruchtfliegen jedoch reicht es bereits aus, normales humanes FUS in Nervenzellen im Überschuss zu produzieren, um die Entwicklung zu stören, die Lebenszeit zu verkürzen und die Beweglichkeit zu beeinträchtigen. Das deutet darauf hin, dass zu viel FUS – auch ohne Mutation – gefährlich ist und eine kontrollierte Systematik bietet, um zu untersuchen, wie und wo diese Gefahr entsteht.
Eine Wendung in der Geschichte der Proteinfehlplatzierung
Das Team erzeugte Fliegen, die eine Version von FUS produzieren, der die eingebaute nukleäre „Postleitzahl“ fehlt, die sogenannte nukleäre Lokalisationsequenz. Ohne dieses Signal gelangt FUS größtenteils nicht in den Kern, sondern reichert sich im Zytoplasma an. Entgegen den Erwartungen waren diese Fliegen weniger krank als solche, die normales FUS produzierten: Sie überlebten besser und entwickelten sich normaler, selbst wenn die Proteinmengen so angepasst wurden, dass beide Versionen in ähnlicher Menge vorhanden waren. Detaillierte Aufnahmen von Larven- und Erwachsenenneuronen bestätigten, dass das Standard-FUS vorwiegend im Zellkern anhäuft, während die veränderte Version größtenteils außerhalb bleibt. Aus diesen Vergleichen schließen die Autorinnen und Autoren, dass FUS in diesem Modell Neurone hauptsächlich über seine Wirkung im Kern schädigt und nicht durch zytoplasmatische Klumpen.
Versteckte Klumpen im Kontrollraum der Zelle
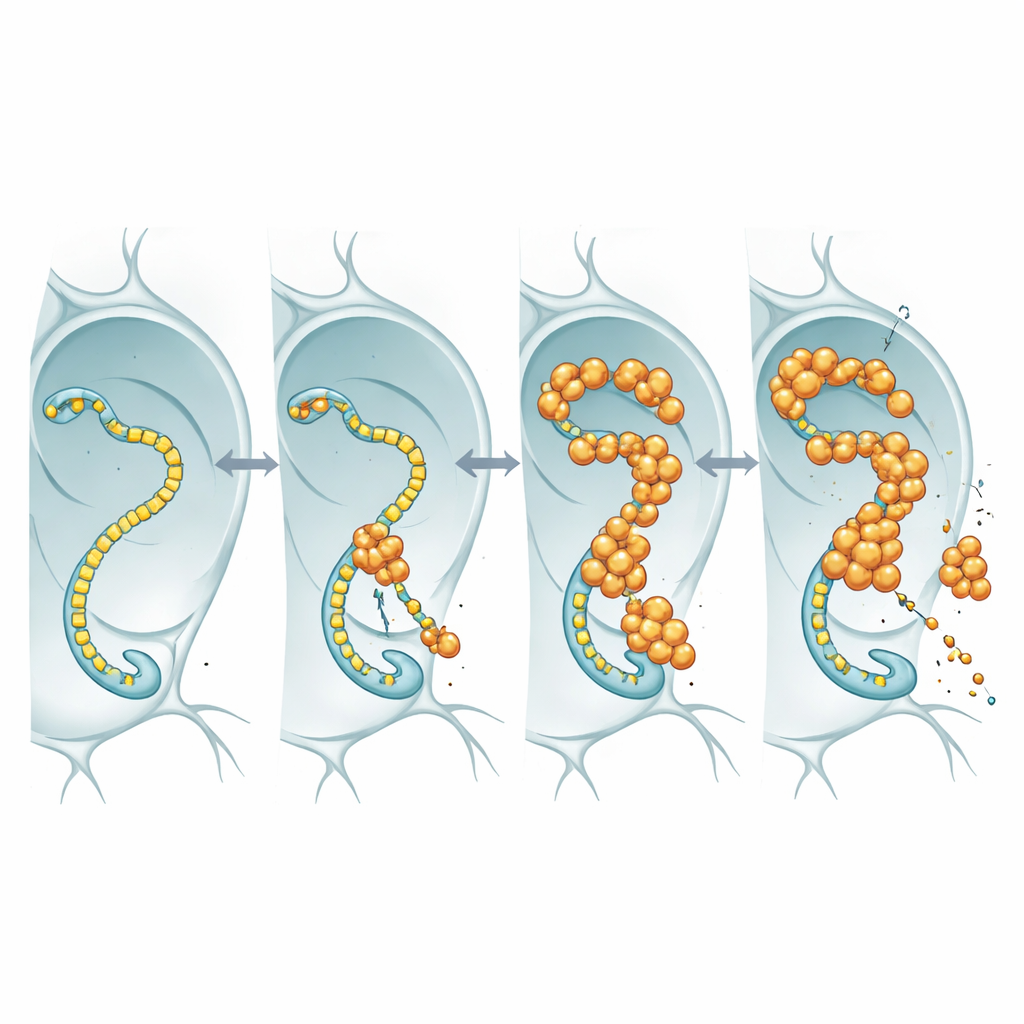
Um zu sehen, was nukleäres FUS tatsächlich macht, versiehten die Forschenden FUS mit einem fluoreszierenden Marker und beobachteten es in den übergroßen Kernen der Speicheldrüsen von Fliegen sowie in Neuronen. Statt harte, unlösliche Aggregate zu bilden, sammelte sich FUS zu hellen, punktförmigen Granula, die dynamisch blieben: Wurde eine Stelle mit einem Laser gebleicht, strömten innerhalb von Sekunden unverbleichte Moleküle zurück. Diese Tropfen traten häufig in Bereichen auf, die ebenfalls hohe Konzentrationen der RNA-Polymerase II enthielten, dem Enzym, das entlang der DNA wandert, um RNA-Kopien von Genen zu erstellen. Die örtliche Nähe deutete darauf hin, dass FUS die Kerntranskriptionsmaschinerie stören könnte, die Neuronen mit den benötigten RNA-Boten versorgt.
Wenn die Genablesemaschine zum Komplizen wird
Die RNA-Polymerase II trägt einen flexiblen Schwanz aus wiederholten kurzen Motiven; dieser Schwanz dient als Landezone für viele regulatorische Proteine, darunter FUS und verwandte Proteine. Mit Fliegenlinien, in denen dieser Schwanz so verändert wurde, dass er weniger oder mehr Wiederholungen enthält, prüfte das Team, ob eine Veränderung der Schwanzlänge die FUS-Toxizität beeinflusst. Bei Fliegen mit FUS-Überproduktion führte eine Verkürzung des Schwanzes zu längerer Lebensdauer, obwohl FUS-mRNA- und Proteinmengen ähnlich blieben. Diese Abhängigkeit von der Länge stimmt mit früheren biochemischen Arbeiten überein, die zeigen, dass längere Schwänze FUS stärker binden. Die einfachste Interpretation ist, dass FUS seine toxische Wirkung ausübt, indem es an diesen Schwanz bindet: Ist der Schwanz lang, bindet FUS stärker, stört die normale Transkriptionskontrolle und Neurone leiden; wird der Schwanz verkürzt, hat FUS weniger Ansatzpunkte und wird weniger schädlich.
Anhaltspunkte aus menschlichem Hirngewebe
Die Autorinnen und Autoren richteten ihren Blick dann auf menschliche postmortale Proben. Im Rückenmarkgewebe von ALS-Patienten mit FUS-Mutationen sahen sie die erwarteten FUS-positiven Einschlusskörperchen in Neuronen, doch die große Untereinheit des menschlichen Enzyms (POLR2A) blieb in ihrem üblichen nukleären Standort. Im Gegensatz dazu zeigten Proben des Frontalkortex von Patienten mit FTLD, die durch FUS-positive Einschlusskörperchen gekennzeichnet sind, häufig abnorme zytoplasmatische Punkte von POLR2A, die manchmal mit FUS-Ablagerungen überlappten. Diese Fehlplatzierungen waren in Kontrollgehirnen nicht vorhanden. Dieses Muster legt nahe, dass in FTLD, aber nicht unbedingt in FUS-ALS, FUS die RNA-Polymerase II aus ihrer Position ziehen könnte und damit die Genexpression in betroffenen Neuronen beeinträchtigt.
Was das für künftige Therapien bedeutet
Insgesamt spricht die Arbeit dafür, dass in diesen Fliegenmodellen die gefährlichste Form von FUS nicht der zytoplasmatische Klumpenbefund ist, sondern die nukleäre Variante, die mit der Genablesemaschinerie interagiert. Indem es dynamische Granula in der Nähe der RNA-Polymerase II bildet und an deren flexiblen Schwanz bindet, scheint überschüssiges FUS den Fluss genetischer Informationen zu stören, die für das Überleben von Neuronen nötig sind. Die Beobachtung, dass POLR2A im menschlichen FTLD-Gehirngewebe ebenfalls fehlplatziert ist, untermauert die Annahme, dass diese nukleäre Partnerschaft zur Krankheit beiträgt. Therapien, die die Ansammlung von FUS im Kern begrenzen oder seine Bindung an die RNA-Polymerase II schwächen, könnten daher Neurone schützen und bieten einen neuen Ansatzpunkt gegen ALS-assoziierte und FUS-positive Demenzen.
Zitation: Moens, T.G., Biasetti, L., Scheveneels, W. et al. Human FUS is toxic via association with RNA polymerase II in Drosophila. Cell Death Dis 17, 310 (2026). https://doi.org/10.1038/s41419-026-08539-x
Schlüsselwörter: FUS-Protein, RNA-Polymerase II, ALS, frontotemporale Demenz, Drosophila-Modell