Clear Sky Science · en
Human FUS is toxic via association with RNA polymerase II in Drosophila
Why this matters for brain health
ALS (Lou Gehrig’s disease) and certain forms of dementia slowly destroy nerve cells, yet we still do not fully understand why those cells die. This study uses fruit flies to probe a protein called FUS, which is linked to some inherited ALS cases and also appears in clumps in a form of dementia called frontotemporal lobar degeneration (FTLD). By asking where in the cell FUS does the most harm, and which partners it interacts with, the researchers uncover a surprising nuclear route to toxicity that may help explain why neurons fail in specific brain diseases.
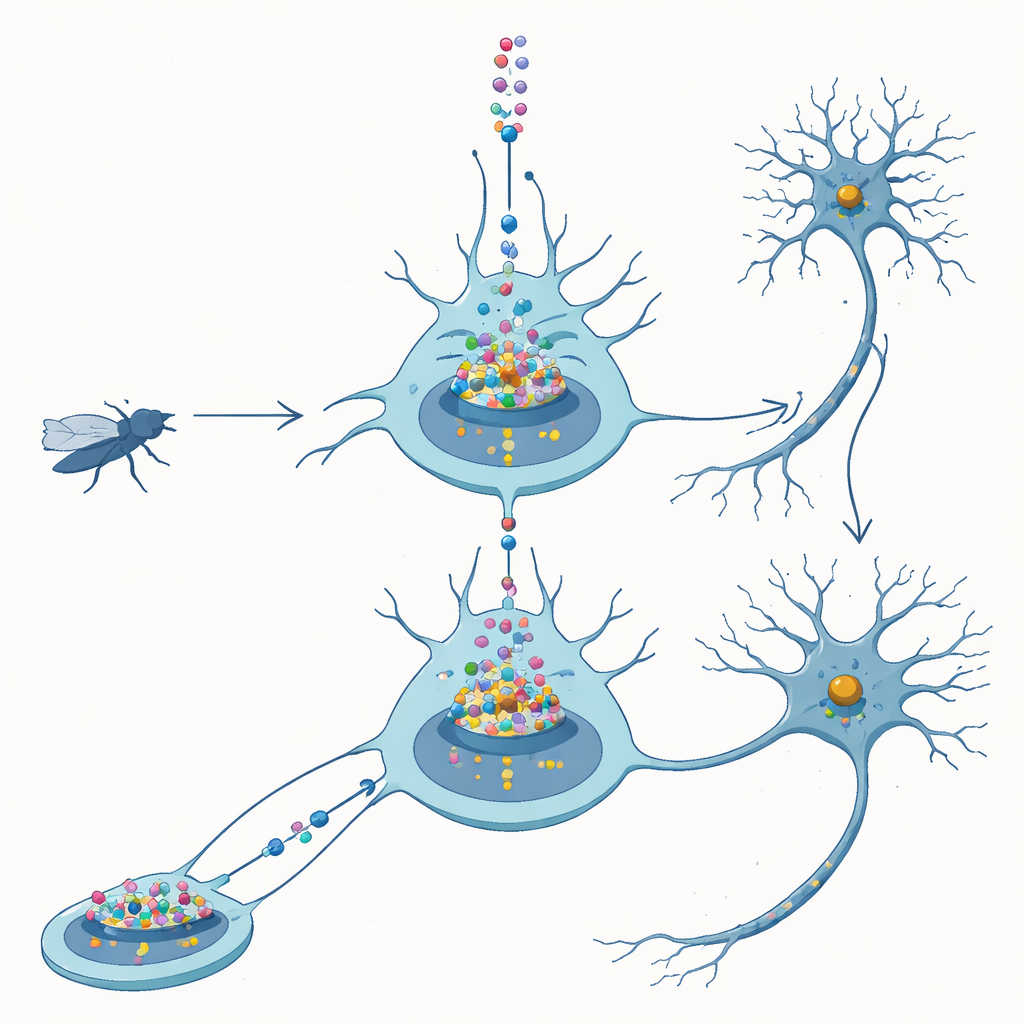
From fly nerves to human disease
FUS is a protein that normally lives in the cell nucleus, where DNA is stored and genes are read out. In patients with FUS-linked ALS, mutant FUS often moves out into the surrounding cytoplasm, forming visible clumps. One popular idea has been that these cytoplasmic deposits are what kill neurons. In fruit flies, however, simply making extra normal human FUS in nerve cells is already enough to block proper development, shorten lifespan, and damage movement. This suggests that too much FUS, even without mutation, is dangerous and provides a controlled system to tease apart how and where that danger arises.
A twist in the tale of protein misplacement
The team engineered flies to produce a version of FUS that lacks its built-in nuclear “postal code,” known as the nuclear localization sequence. Without this tag, FUS largely fails to enter the nucleus and instead builds up in the cytoplasm. Counter to expectations, these flies were less sick than flies making normal FUS: they survived better and developed more normally, even when protein levels were adjusted so the two versions were present at similar amounts. Detailed imaging of larval and adult neurons confirmed that the standard FUS piled up mainly in the nucleus, whereas the altered version stayed mostly outside. These comparisons led the authors to conclude that FUS in this model harms neurons primarily through what it does in the nucleus, not through cytoplasmic clumps.
Hidden clumps in the cell’s control room
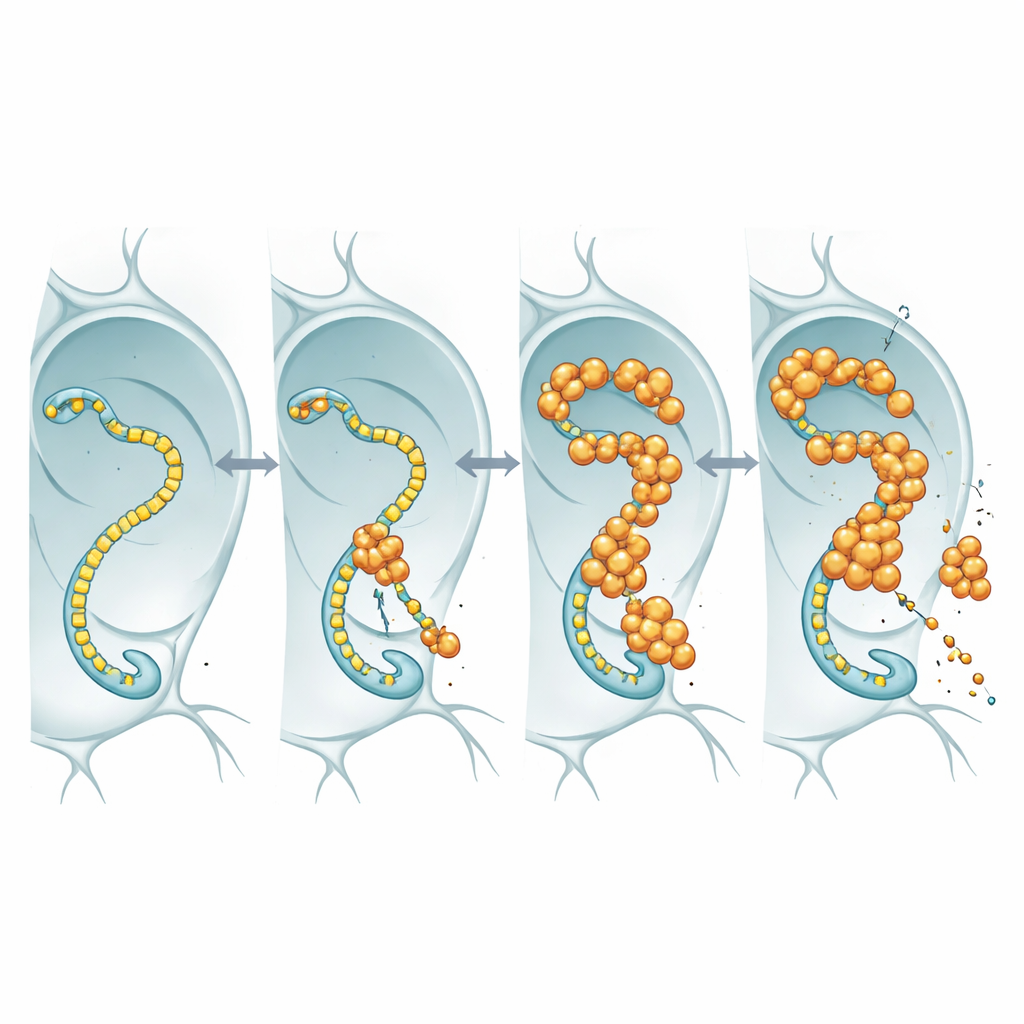
To see what nuclear FUS is actually doing, the researchers tagged FUS with a fluorescent marker and watched it in the oversized nuclei of fly salivary glands and in neurons. Rather than forming hard, insoluble aggregates, FUS gathered into bright, punctate granules that remained dynamic: when a patch was bleached with a laser, unbleached molecules flowed back in within seconds. These droplets tended to appear in regions that also contained high levels of RNA polymerase II, the enzyme that travels along DNA to make RNA copies of genes. This colocation hinted that FUS might be interfering with the core transcription machinery that keeps neurons supplied with the RNA messages they need.
When the gene-reading machine becomes a partner in crime
RNA polymerase II carries a flexible tail made of repeated short motifs; this tail acts as a landing strip for many regulatory proteins, including FUS and its relatives. Using fly lines in which this tail was engineered to contain fewer or more repeats, the team asked whether changing tail length would alter FUS toxicity. In flies overproducing FUS, shortening the tail made the animals live longer, even though FUS RNA and protein levels stayed similar. This length dependence matches earlier biochemical work showing that longer tails bind FUS more avidly. The simplest interpretation is that FUS exerts its toxic effect by engaging this tail: when the tail is long, FUS binds more strongly, disrupts normal transcriptional control, and neurons suffer; when the tail is shortened, FUS has less to grab onto and becomes less harmful.
Clues from human brain tissue
The authors then turned to human post-mortem samples. In spinal cord tissue from ALS patients carrying FUS mutations, they saw the expected FUS-positive inclusions in neurons, but the human version of the enzyme’s large subunit (POLR2A) remained in its usual nuclear location. By contrast, in frontal cortex from patients with FTLD marked by FUS-positive inclusions, POLR2A itself often appeared in abnormal cytoplasmic spots that sometimes overlapped with FUS deposits. These misplacements were absent in control brains. This pattern suggests that in FTLD, but not necessarily in FUS-ALS, FUS may drag RNA polymerase II out of position, potentially crippling gene expression in affected neurons.
What this means for future therapies
Taken together, the work argues that in these fly models, the most dangerous form of FUS is not the cytoplasmic clump but the nuclear version that interacts with the gene-reading machinery. By forming dynamic granules near RNA polymerase II and binding its flexible tail, excess FUS appears to disturb the flow of genetic information needed to keep neurons alive. The observation that POLR2A also mislocalises in human FTLD brain tissue strengthens the case that this nuclear partnership contributes to disease. Therapies that limit FUS buildup in the nucleus or weaken its grip on RNA polymerase II might therefore protect neurons, offering a new angle of attack on ALS-related and FUS-positive dementias.
Citation: Moens, T.G., Biasetti, L., Scheveneels, W. et al. Human FUS is toxic via association with RNA polymerase II in Drosophila. Cell Death Dis 17, 310 (2026). https://doi.org/10.1038/s41419-026-08539-x
Keywords: FUS protein, RNA polymerase II, ALS, frontotemporal dementia, Drosophila model