Clear Sky Science · fr
Le FUS humain est toxique via son association avec l’ARN polymérase II chez Drosophila
Pourquoi cela compte pour la santé cérébrale
La SLA (maladie de Lou Gehrig) et certaines formes de démence détruisent lentement les cellules nerveuses, mais nous ne comprenons toujours pas entièrement pourquoi ces cellules meurent. Cette étude utilise la mouche du vinaigre pour étudier une protéine appelée FUS, liée à certains cas héréditaires de SLA et qui apparaît aussi en agrégats dans une forme de démence appelée dégénérescence lobaire frontotemporale (DLFT). En cherchant où dans la cellule le FUS est le plus nocif et avec quels partenaires il interagit, les chercheurs mettent au jour une voie nucléaire surprenante de toxicité qui pourrait aider à expliquer pourquoi les neurones échouent dans des maladies cérébrales spécifiques.
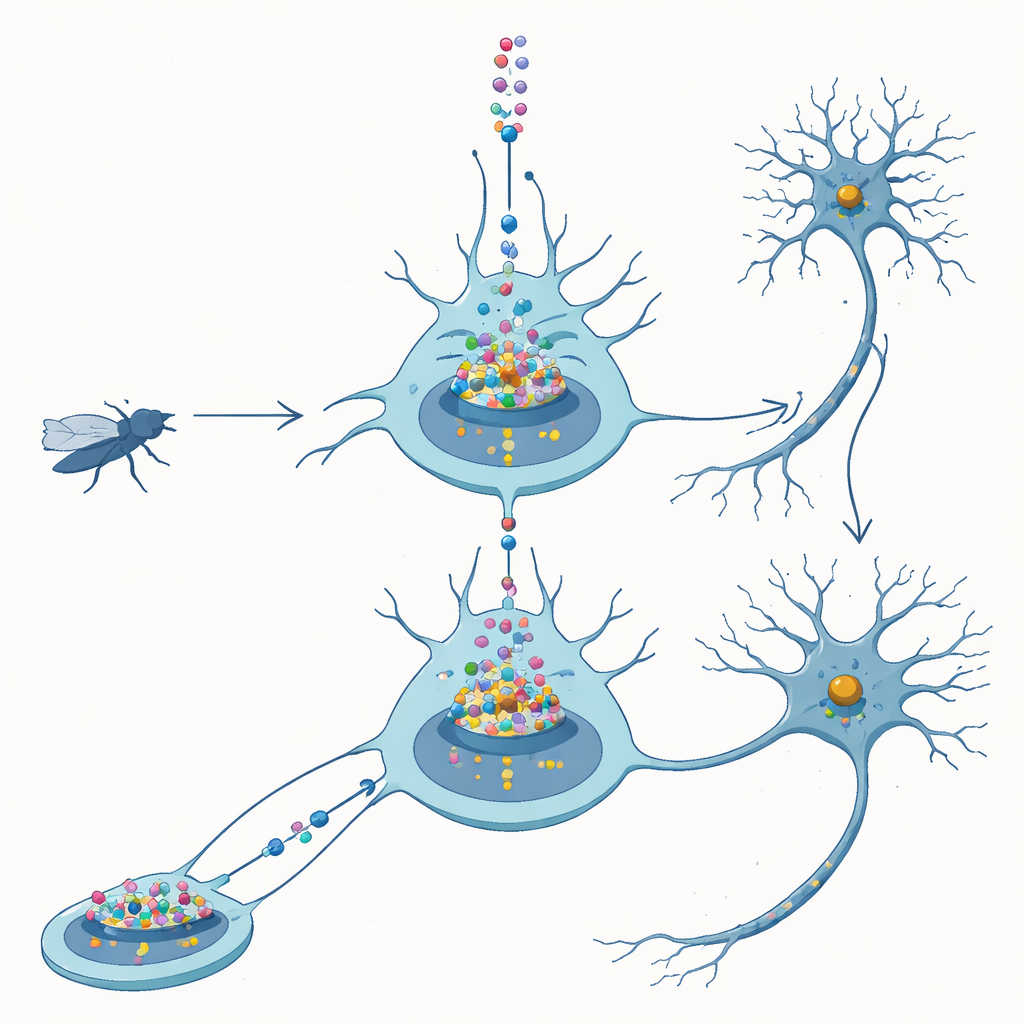
Des nerfs de la mouche à la maladie humaine
FUS est une protéine qui vit normalement dans le noyau cellulaire, où l’ADN est stocké et où les gènes sont lus. Chez les patients porteurs de mutations FUS liées à la SLA, le FUS mutant migre souvent vers le cytoplasme environnant et forme des agrégats visibles. Une idée répandue est que ces dépôts cytoplasmiques tuent les neurones. Chez la drosophile, toutefois, le simple fait de produire en excès le FUS humain normal dans les cellules nerveuses suffit déjà à bloquer un développement correct, raccourcir la durée de vie et altérer la motricité. Cela suggère qu’un excès de FUS, même sans mutation, est dangereux et fournit un système contrôlé pour démêler comment et où ce danger se manifeste.
Un retournement dans l’histoire du mauvais placement des protéines
L’équipe a conçu des mouches pour produire une version de FUS dépourvue de son « code postal » nucléaire, la séquence de localisation nucléaire. Sans cette étiquette, le FUS entre en grande partie en défaut dans le noyau et s’accumule plutôt dans le cytoplasme. Contrairement aux attentes, ces mouches étaient moins malades que celles produisant le FUS normal : elles survivaient mieux et se développaient plus normalement, même lorsque les niveaux de protéine étaient ajustés pour que les deux versions soient présentes à quantités comparables. Des images détaillées des neurones larvaires et adultes ont confirmé que le FUS standard s’accumulait principalement dans le noyau, tandis que la version altérée restait majoritairement à l’extérieur. Ces comparaisons ont conduit les auteurs à conclure que, dans ce modèle, le FUS nuit aux neurones principalement par son action dans le noyau, et non par des agrégats cytoplasmiques.
Agrégats cachés dans la salle de contrôle de la cellule
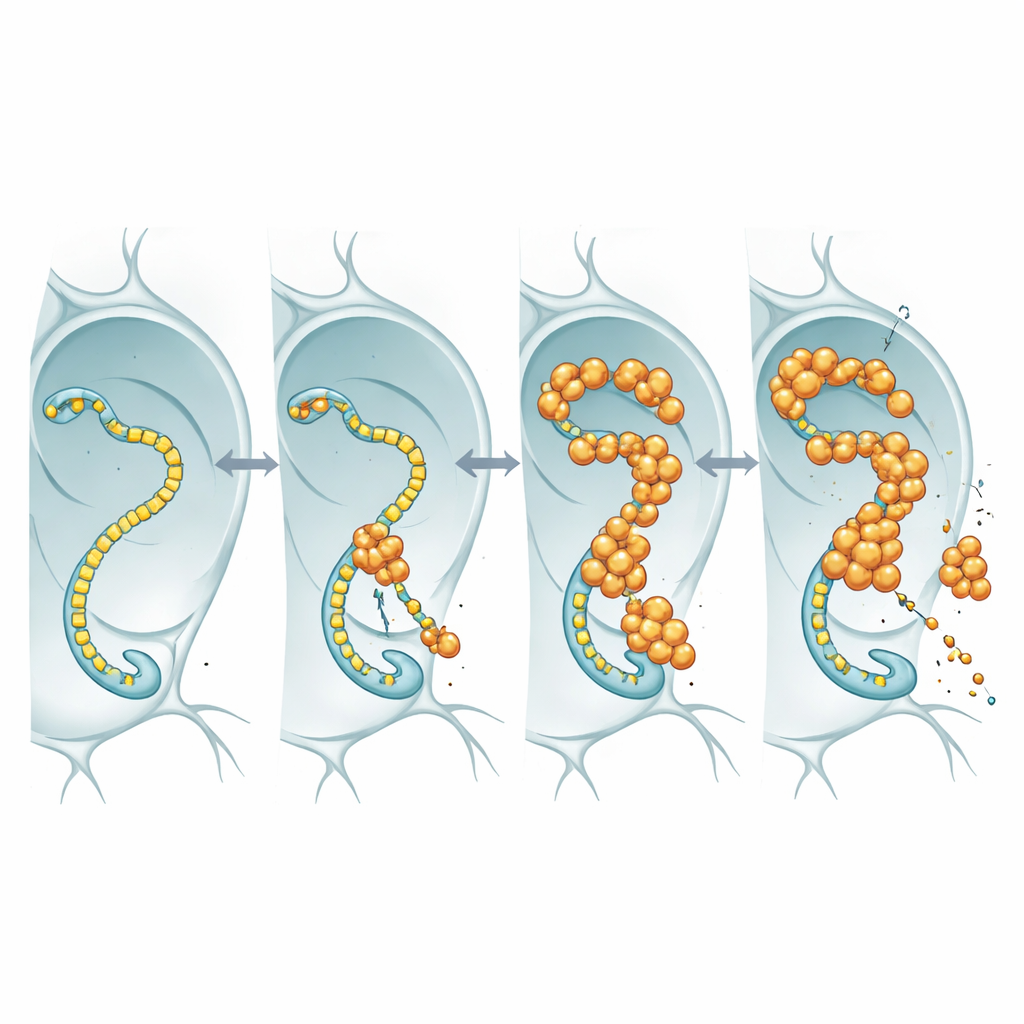
Pour voir ce que fait réellement le FUS nucléaire, les chercheurs ont marqué FUS par un traceur fluorescent et l’ont observé dans les noyaux surdimensionnés des glandes salivaires de la mouche et dans les neurones. Plutôt que de former des agrégats durs et insolubles, le FUS se rassemblait en granules ponctués et lumineux qui restaient dynamiques : lorsqu’une zone était photo-bleachée au laser, des molécules non-brûlées revenaient en quelques secondes. Ces gouttelettes avaient tendance à apparaître dans des régions contenant aussi de fortes concentrations d’ARN polymérase II, l’enzyme qui parcourt l’ADN pour fabriquer des copies ARN des gènes. Cette colocalisation suggère que le FUS pourrait interférer avec la machinerie de transcription centrale qui fournit aux neurones les messages ARN dont ils ont besoin.
Quand la machine de lecture des gènes devient complice
L’ARN polymérase II possède une queue flexible composée de motifs courts répétés ; cette queue sert de piste d’atterrissage pour de nombreuses protéines régulatrices, y compris FUS et ses apparentés. En utilisant des lignées de mouches dans lesquelles cette queue a été conçue pour contenir moins ou plus de répétitions, l’équipe a testé si changer la longueur de la queue modifierait la toxicité du FUS. Chez les mouches surexprimant le FUS, raccourcir la queue a prolongé la durée de vie des animaux, même si les niveaux d’ARN et de protéine FUS restaient similaires. Cette dépendance à la longueur correspond à des travaux biochimiques antérieurs montrant que les queues plus longues lient le FUS de manière plus avides. L’interprétation la plus simple est que le FUS exerce son effet toxique en s’engageant sur cette queue : lorsque la queue est longue, FUS se lie plus fortement, perturbe le contrôle transcriptionnel normal et les neurones souffrent ; lorsque la queue est raccourcie, il y a moins de prise pour FUS et il devient moins nocif.
Indices provenant de tissus cérébraux humains
Les auteurs se sont ensuite tournés vers des échantillons post-mortem humains. Dans des tissus de moelle épinière de patients SLA porteurs de mutations FUS, ils ont observé les inclusions positives pour FUS attendues dans les neurones, mais la grande sous-unité de l’enzyme humaine (POLR2A) restait à sa localisation nucléaire habituelle. En revanche, dans le cortex frontal de patients atteints de DLFT marqué par des inclusions positives pour FUS, POLR2A apparaissait souvent en points cytoplasmiques anormaux qui chevauchaient parfois les dépôts de FUS. Ces mauvais placements étaient absents dans les cerveaux témoins. Ce schéma suggère que dans la DLFT, mais pas nécessairement dans la SLA liée à FUS, FUS pourrait entraîner l’ARN polymérase II hors de sa position, affaiblissant potentiellement l’expression génique dans les neurones affectés.
Ce que cela implique pour les thérapies futures
Pris ensemble, les résultats soutiennent que, dans ces modèles de mouche, la forme la plus dangereuse du FUS n’est pas l’amas cytoplasmique mais la version nucléaire qui interagit avec la machinerie de lecture des gènes. En formant des granules dynamiques à proximité de l’ARN polymérase II et en se liant à sa queue flexible, l’excès de FUS semble perturber le flux d’information génétique nécessaire au maintien des neurones. L’observation que POLR2A se relocalise aussi dans des tissus cérébraux humains de DLFT renforce l’hypothèse que ce partenariat nucléaire contribue à la maladie. Des thérapies qui limiteraient l’accumulation de FUS dans le noyau ou affaibliraient son ancrage à l’ARN polymérase II pourraient donc protéger les neurones, offrant un nouvel angle d’attaque contre la SLA et les démences FUS-positives.
Citation: Moens, T.G., Biasetti, L., Scheveneels, W. et al. Human FUS is toxic via association with RNA polymerase II in Drosophila. Cell Death Dis 17, 310 (2026). https://doi.org/10.1038/s41419-026-08539-x
Mots-clés: Protéine FUS, ARN polymérase II, SLA, démence frontotemporale, Modèle Drosophila