Clear Sky Science · nl
Hybride selectiemodel voor kenmerken en classificatie met behulp van hoog-dimensionale gegevens gebaseerd op een metaheuristische algoritme voor de diagnose van hersenkanker
Waarom slimmere tests voor hersentumoren ertoe doen
Hersentumoren behoren tot de dodelijkste kankers, maar artsen hebben nog steeds moeite om verschillende tumortypes snel en nauwkeurig te onderscheiden. Traditionele laboratoriumtests kunnen traag zijn, en moderne gengebaseerde tests produceren een overweldigende stroom data: tienduizenden genmetingen per patiënt. Deze studie beschrijft een computergebaseerde methode die door dit genetische lawaai heen siebt om een kleine set sleutelgenen te vinden die helpen gevaarlijke tumoren te onderscheiden van minder agressieve gevallen, met als doel nauwkeurigere diagnoses en uiteindelijk betere behandelbeslissingen te ondersteunen.
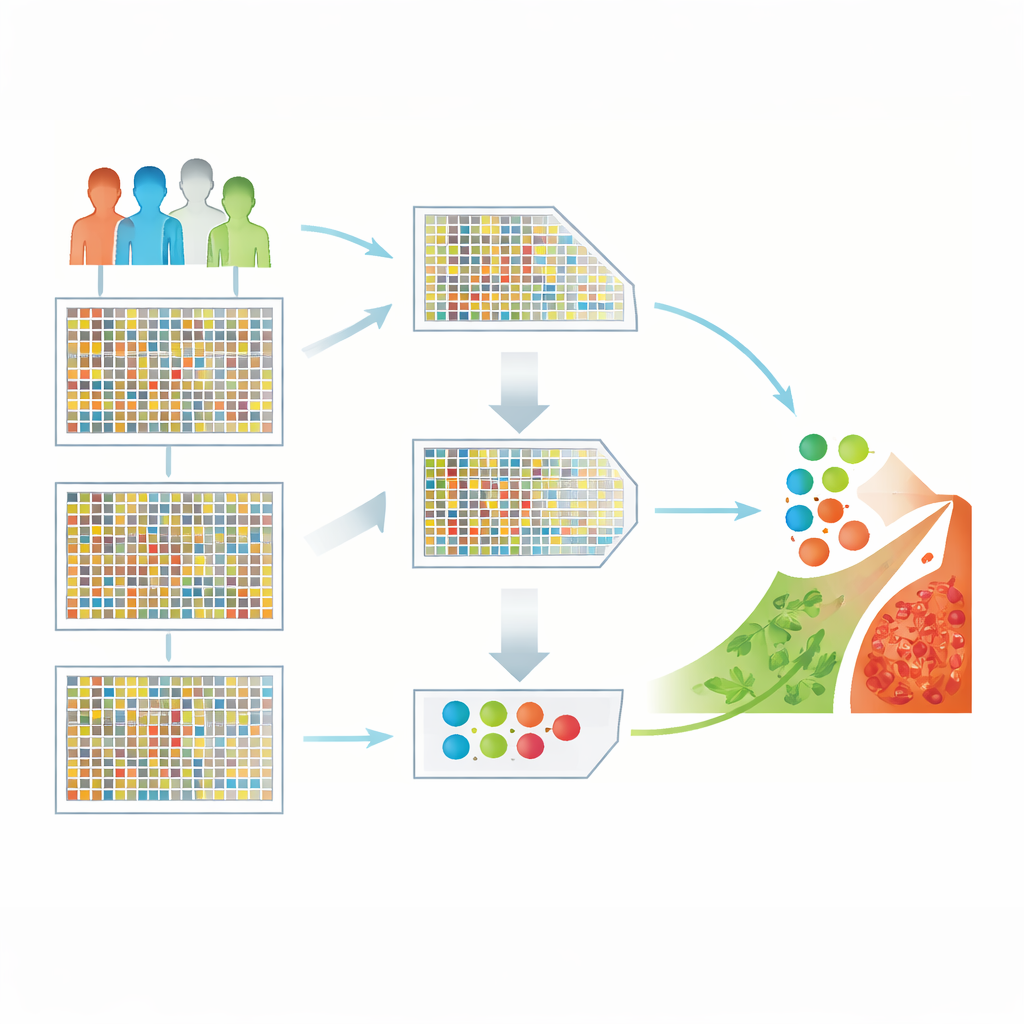
Een zee van genen omzetten in bruikbare aanwijzingen
De onderzoekers werkten met een openbaar dataset voor hersenkanker met 130 patiëntmonsters, elk gemeten op meer dan 54.000 genen. Dergelijke hoog-dimensionale gegevens zijn krachtig maar lastig: er zijn veel meer genmetingen dan patiënten, en veel genen zijn irrelevant of redundant. Als je deze direct in een leeralgoritme stopt, kan die onbalans leiden tot misleidende patronen en slechte prestaties bij nieuwe patiënten. De centrale uitdaging is alleen de meest informatieve genen te behouden en de rest weg te gooien, zonder belangrijke biologische signalen te verliezen.
Een tweestapsmethode om de meest zeggende genen te vinden
Om dit aan te pakken, ontwierp het team een hybride pipeline voor kenmerkselectie. In de eerste stap scant een snelle statistische filter, genoemd “minimum redundantie, maximale relevantie” (mRMR), alle genen en behoudt diegenen die sterk verbonden zijn met hersenkanker en tegelijkertijd zo verschillend mogelijk van elkaar zijn. Dit brengt de genlijst snel terug van tienduizenden tot een beter hanteerbare set. In de tweede stap behandelt een zoekmethode geïnspireerd op het jachtgedrag van roofvogels—Harris Hawks Optimization—elke mogelijke subsets van genen als een kandidaatoplossing en “jaagt” iteratief op combinaties die de beste classificatieresultaten geven. Samen reduceren deze stappen de oorspronkelijke 54.676 genen tot slechts 50 die nog steeds de kern van de ziekte vastleggen.
Machines leren een scherpere diagnostische grens te trekken
Zodra een compacte geneset was geïdentificeerd, trainden de auteurs meerdere machine-learningmodellen om te onderscheiden tussen vijf hersenkankercategorieën, en meer algemeen tussen kwaadaardig en minder gevaarlijk weefsel. Een belangrijk model, de support vector machine, heeft een paar gevoeligheidsknoppen die zijn gedrag sterk beïnvloeden. In plaats van deze bij toeval af te stellen, gebruikte het team drie optimalisatiestrategieën—Particle Swarm Optimization, Differential Evolution en Harris Hawks Optimization—om systematisch naar de beste instellingen te zoeken. Ze beoordeelden de prestaties met rigoureuze, herhaalde cross-validatie en aanvullende controles zoals bootstrap-resampling en leercurve-analyse om zeker te zijn dat de modellen niet simpelweg het kleine dataset memoriseren.
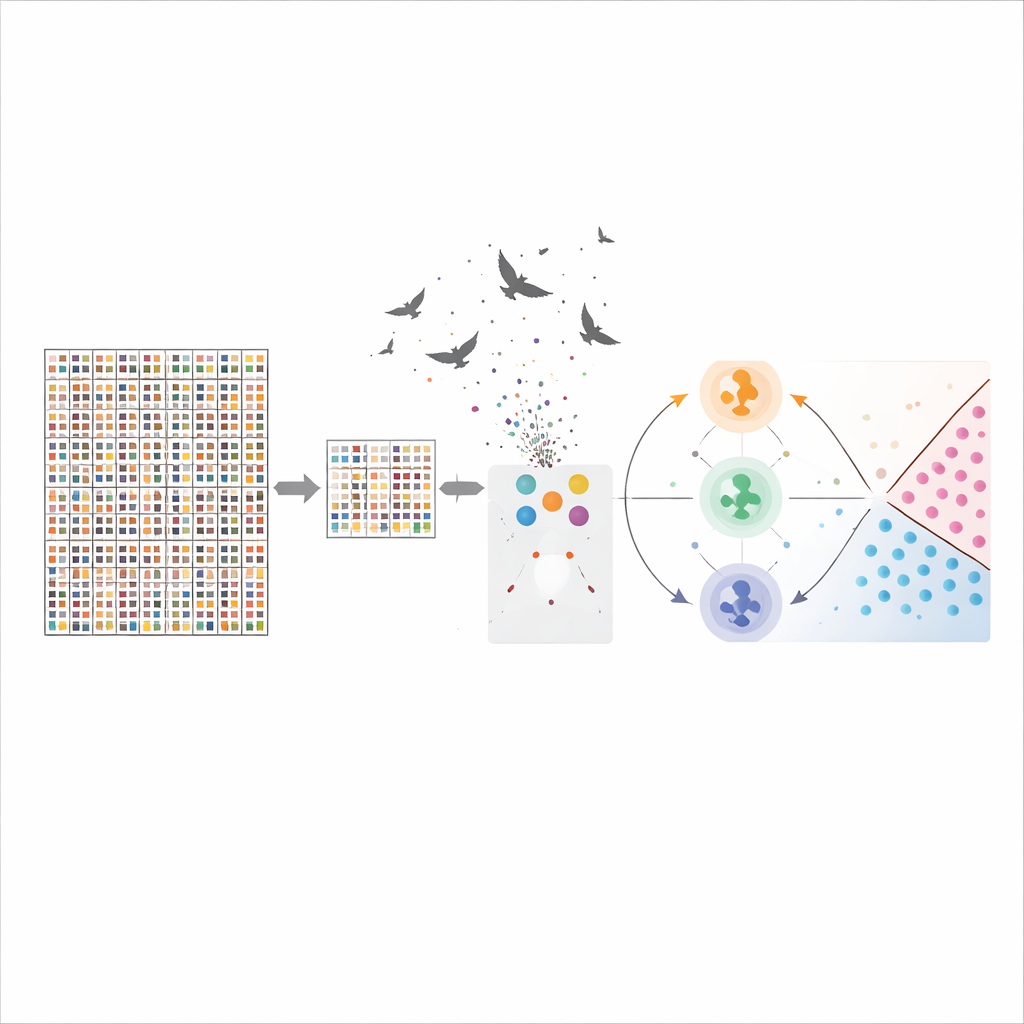
Hoe goed het systeem werkt en wat de genen betekenen
Met de hybride genselectie en optimalisatie behaalde de support vector machine een nauwkeurigheid van ongeveer 96% op de hersenkankergegevens, wat duidelijk beter presteerde dan meer conventionele methoden zoals beslisbomen. De methode drukte ook het aantal genen terug van meer dan 54.000 naar 50, terwijl de voorspellende kracht behouden bleef en zelfs verbeterde. Veel van de geselecteerde genen zijn al bekend betrokken te zijn bij celgroei, DNA-herstel, bloedvaatvorming of immuunrespons in hersentumoren, wat biologische geloofwaardigheid geeft aan de keuzes van de computer. Dit betekent dat het model niet alleen goed is in classificatie, maar onderzoekers ook wijst op potentiële biomarkers die de moeite van verder laboratoriumonderzoek waard zijn.
Wat dit voor patiënten zou kunnen betekenen
In eenvoudige bewoordingen laat de studie zien dat het mogelijk is een enorme hoeveelheid genetische data samen te persen tot een kleine, betekenisvolle gensignatuur die helpt verschillende hersentumortypes met hoge betrouwbaarheid te scheiden. Hoewel het werk nog gebaseerd is op een relatief kleine patiëntengroep en moet worden getest op grotere, meer diverse populaties, suggereert het een route naar snellere, gengebaseerde diagnostische hulpmiddelen die zowel nauwkeurig als interpreteerbaar zijn. Als ze worden gevalideerd en geïntegreerd in klinische workflows, zouden dergelijke hulpmiddelen artsen sterkere onderbouwing kunnen geven bij het kiezen van behandelingen en wetenschappers helpen zich te concentreren op een korte lijst genen die waarschijnlijk invloed hebben op hoe hersenkankers ontstaan, groeien en reageren op therapie.
Bronvermelding: Manhrawy, I.I.M., Fathi, H., Alsekait, D.M. et al. Hybrid feature selection and classification model using high-dimensional data based on a metaheuristic algorithm for brain cancer diagnosis. Sci Rep 16, 11909 (2026). https://doi.org/10.1038/s41598-026-41573-5
Trefwoorden: diagnose van hersenkanker, genexpressie, kenmerkselectie, machine learning, biomarkers