Clear Sky Science · en
Hybrid feature selection and classification model using high-dimensional data based on a metaheuristic algorithm for brain cancer diagnosis
Why smarter tests for brain tumors matter
Brain tumors are among the deadliest cancers, yet doctors still struggle to distinguish different tumor types quickly and accurately. Traditional lab tests can be slow, and modern gene-based tests generate an overwhelming flood of data: tens of thousands of gene measurements for each patient. This study presents a computer-based method that sifts through this genetic noise to find a small set of key genes that help separate dangerous tumors from less aggressive cases, aiming to support more precise diagnosis and, eventually, better treatment decisions.
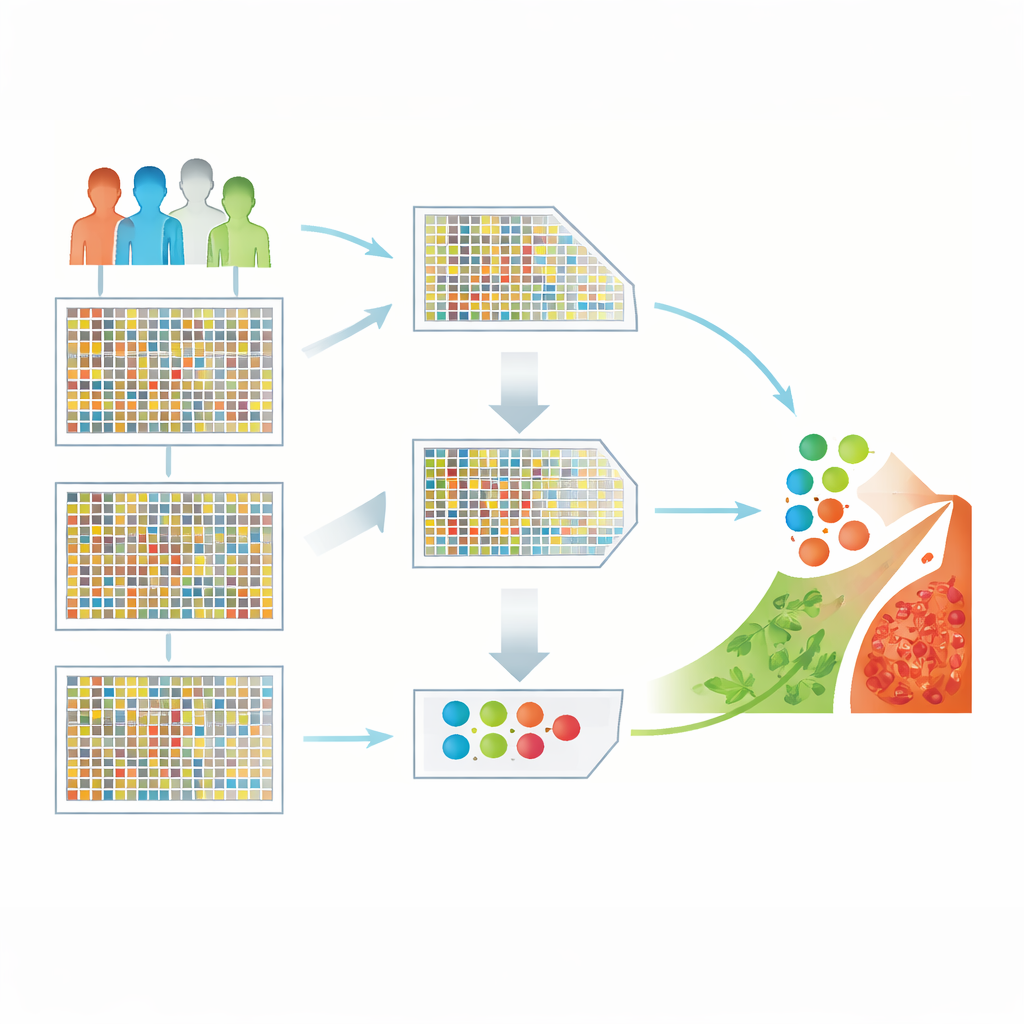
Turning a sea of genes into usable clues
The researchers worked with a public brain cancer dataset containing 130 patient samples, each measured for more than 54,000 genes. Such high-dimensional data are powerful but tricky: there are far more gene measurements than patients, and many genes are irrelevant or redundant. If fed directly into a learning algorithm, this imbalance can lead to misleading patterns and poor performance on new patients. The central challenge is to keep only the most informative genes while throwing away the rest, without losing important biological signals.
A two-step way to find the most telling genes
To tackle this, the team designed a hybrid feature selection pipeline. In the first step, a fast statistical filter called “minimum redundancy, maximum relevance” (mRMR) scans all genes and keeps those that are strongly linked to brain cancer while being as different from each other as possible. This quickly shrinks the gene list from tens of thousands to a more manageable set. In the second step, a search method inspired by the hunting behavior of birds of prey—Harris Hawks Optimization—treats each possible gene subset as a candidate solution and iteratively “hunts” for combinations that give the best classification results. Together, these stages reduce the original 54,676 genes down to only 50 that still capture the essence of the disease.
Teaching machines to draw a sharper diagnostic line
Once a compact gene set was identified, the authors trained several machine-learning models to distinguish among five brain cancer categories, and more broadly between malignant and less dangerous tissue. A key model, the support vector machine, needs a couple of sensitivity knobs that strongly affect its behavior. Rather than tuning these by trial and error, the team used three optimization strategies—Particle Swarm Optimization, Differential Evolution, and Harris Hawks Optimization—to systematically search for the best settings. They evaluated performance using rigorous, repeated cross-validation and additional checks such as bootstrap resampling and learning-curve analysis to ensure that the models were not simply memorizing the small dataset.
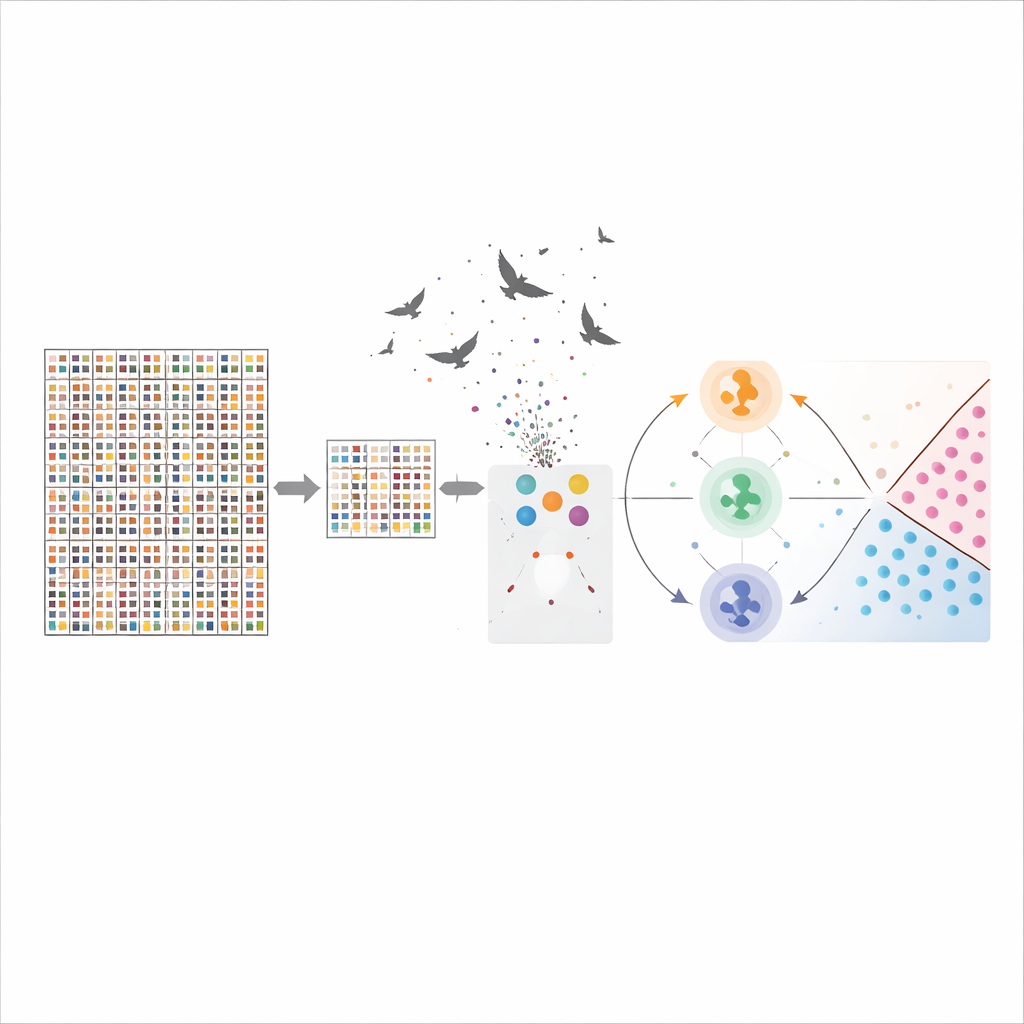
How well the system works and what the genes mean
With the hybrid gene selection and optimization in place, the support vector machine achieved an accuracy of about 96% on the brain cancer data, clearly outperforming more conventional methods such as decision trees. The method also pushed the number of genes down from over 54,000 to 50 while preserving, and even improving, predictive power. Many of the selected genes are already known to be involved in cell growth, DNA repair, blood vessel formation, or immune response in brain tumors, lending biological credibility to the computer’s choices. This means the model is not only good at classification but also points researchers toward potential biomarkers worth studying in the lab.
What this could mean for patients
In plain terms, the study shows that it is possible to compress an enormous volume of genetic data into a small, meaningful gene “signature” that helps separate different brain tumor types with high reliability. While the work is still based on a relatively small patient group and needs to be tested on larger, more diverse populations, it suggests a path toward faster, gene-based diagnostic tools that are both accurate and interpretable. If validated and integrated into clinical workflows, such tools could give doctors stronger evidence when choosing treatments and help scientists focus on a short list of genes most likely to influence how brain cancers start, grow, and respond to therapy.
Citation: Manhrawy, I.I.M., Fathi, H., Alsekait, D.M. et al. Hybrid feature selection and classification model using high-dimensional data based on a metaheuristic algorithm for brain cancer diagnosis. Sci Rep 16, 11909 (2026). https://doi.org/10.1038/s41598-026-41573-5
Keywords: brain cancer diagnosis, gene expression, feature selection, machine learning, biomarkers