Clear Sky Science · it
Selezione di caratteristiche ibrida e modello di classificazione per dati ad alta dimensionalità basato su un algoritmo metaeuristico per la diagnosi del cancro cerebrale
Perché servono test più intelligenti per i tumori cerebrali
I tumori cerebrali sono tra i tumori più letali, eppure i medici faticano ancora a distinguere rapidamente e con precisione i diversi tipi di tumore. I test di laboratorio tradizionali possono essere lenti, e i moderni test basati sui geni generano un flusso di dati schiacciante: decine di migliaia di misurazioni geniche per ciascun paziente. Questo studio presenta un metodo informatico che setaccia questo rumore genetico per trovare un piccolo insieme di geni chiave che aiutano a separare i tumori più pericolosi da quelli meno aggressivi, con l’obiettivo di supportare una diagnosi più precisa e, in prospettiva, decisioni terapeutiche migliori.
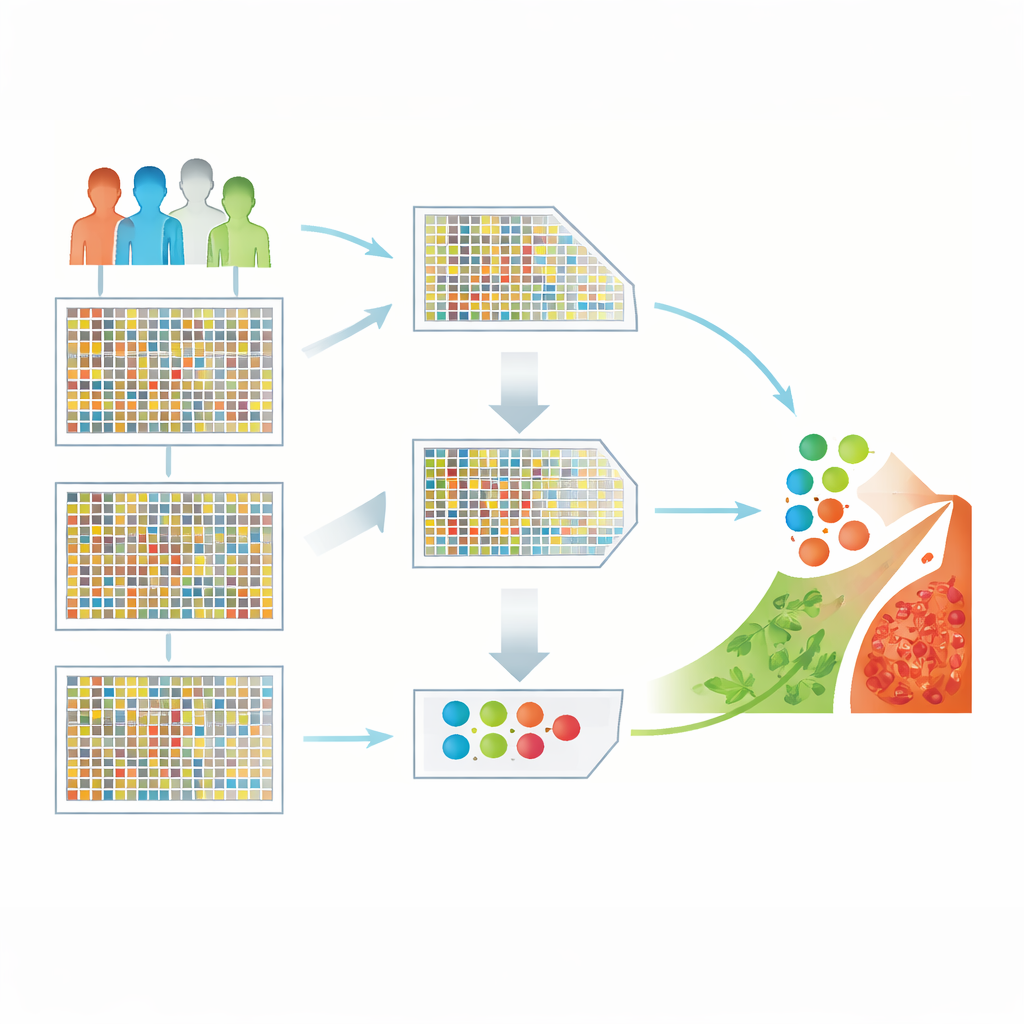
Trasformare un mare di geni in indizi utili
I ricercatori hanno lavorato con un dataset pubblico sul cancro cerebrale contenente 130 campioni di pazienti, ciascuno misurato per più di 54.000 geni. Dati ad alta dimensionalità come questi sono potenti ma complessi: ci sono molte più misurazioni geniche che pazienti, e molti geni sono irrilevanti o ridondanti. Se inseriti direttamente in un algoritmo di apprendimento, questo squilibrio può portare a pattern fuorvianti e scarse prestazioni su nuovi pazienti. La sfida centrale è mantenere solo i geni più informativi scartando il resto, senza perdere segnali biologici importanti.
Un approccio in due fasi per trovare i geni più rivelatori
Per affrontare il problema, il team ha progettato una pipeline ibrida di selezione delle caratteristiche. Nella prima fase, un filtro statistico rapido chiamato «minima ridondanza, massima rilevanza» (mRMR) esamina tutti i geni e mantiene quelli fortemente collegati al cancro cerebrale e al contempo il più differenti possibile l’uno dall’altro. Questo riduce rapidamente la lista da decine di migliaia a un insieme più gestibile. Nella seconda fase, un metodo di ricerca ispirato al comportamento di caccia dei rapaci — l’ottimizzazione Harris Hawks (Harris Hawks Optimization) — tratta ogni possibile sottoinsieme di geni come una soluzione candidata e «caccia» iterativamente combinazioni che producono i migliori risultati di classificazione. Insieme, queste fasi riducono gli originali 54.676 geni a soli 50 che catturano comunque l’essenza della malattia.
Addestrare macchine a tracciare una linea diagnostica più netta
Una volta identificato un set compatto di geni, gli autori hanno addestrato diversi modelli di apprendimento automatico per distinguere cinque categorie di tumore cerebrale e, più in generale, tra tessuto maligno e meno pericoloso. Un modello chiave, la macchina a vettori di supporto (support vector machine), richiede un paio di parametri di sensibilità che influenzano fortemente il suo comportamento. Piuttosto che sintonizzarli per tentativi, il team ha utilizzato tre strategie di ottimizzazione — Particle Swarm Optimization, Differential Evolution e Harris Hawks Optimization — per cercare sistematicamente le migliori impostazioni. Hanno valutato le prestazioni usando una rigorosa validazione incrociata ripetuta e controlli aggiuntivi come il bootstrap resampling e l’analisi delle curve di apprendimento per assicurarsi che i modelli non stessero semplicemente memorizzando il piccolo dataset.
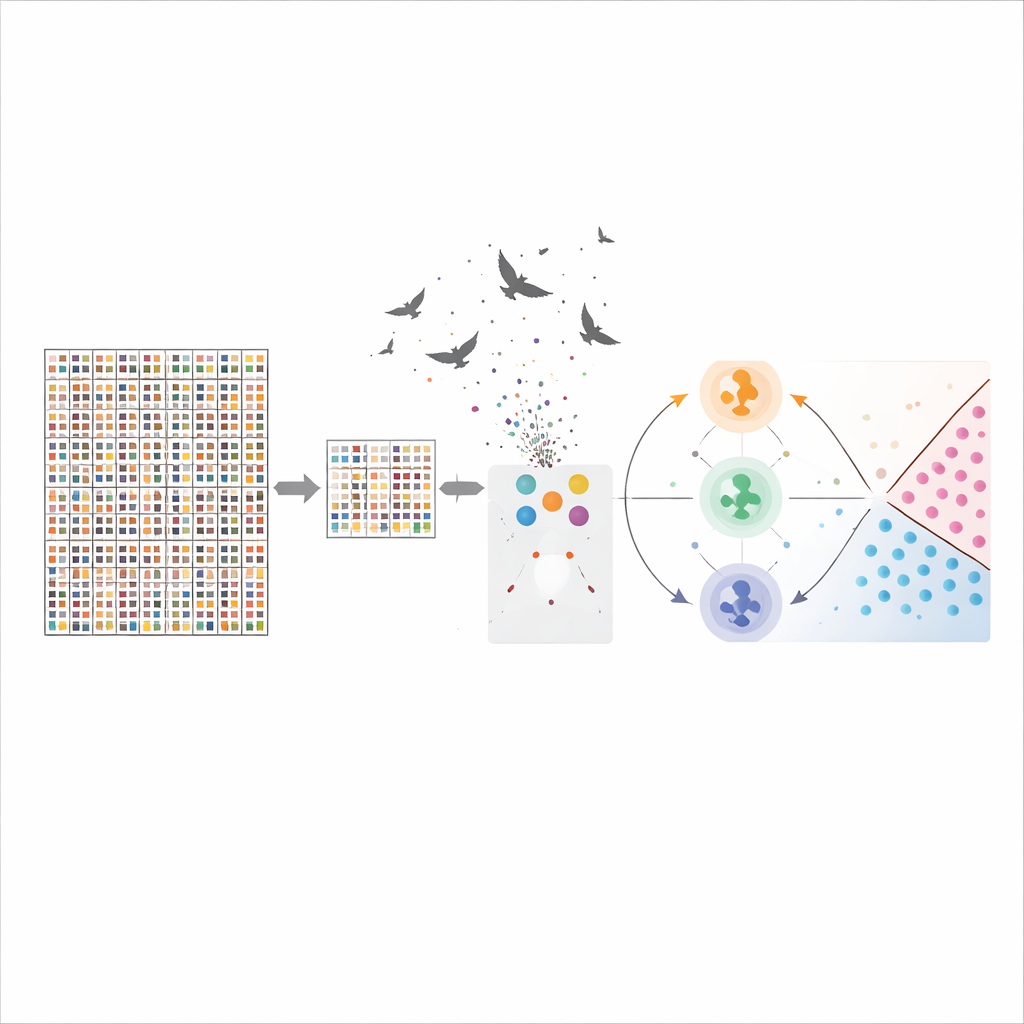
Quanto funziona il sistema e cosa significano i geni
Con la selezione ibrida dei geni e l’ottimizzazione applicata, la macchina a vettori di supporto ha raggiunto un’accuratezza di circa il 96% sui dati sul cancro cerebrale, superando nettamente metodi più convenzionali come gli alberi decisionali. Il metodo ha inoltre ridotto il numero di geni da oltre 54.000 a 50 pur preservando, e perfino migliorando, la potenza predittiva. Molti dei geni selezionati sono già noti per essere coinvolti nella crescita cellulare, nella riparazione del DNA, nella formazione di vasi sanguigni o nella risposta immunitaria nei tumori cerebrali, conferendo credibilità biologica alle scelte del computer. Questo significa che il modello non è solo efficace nella classificazione, ma indirizza anche i ricercatori verso potenziali biomarcatori da studiare in laboratorio.
Cosa potrebbe significare per i pazienti
In termini semplici, lo studio mostra che è possibile comprimere un enorme volume di dati genetici in una piccola «firma» genica significativa che aiuta a separare diversi tipi di tumore cerebrale con alta affidabilità. Anche se il lavoro si basa ancora su un gruppo di pazienti relativamente ridotto e necessita di essere testato su popolazioni più ampie e diversificate, suggerisce una strada verso strumenti diagnostici basati sui geni più rapidi, accurati e interpretabili. Se validati e integrati nei percorsi clinici, tali strumenti potrebbero fornire ai medici prove più solide nella scelta delle terapie e aiutare gli scienziati a concentrarsi su una breve lista di geni più propensi a influenzare l’insorgenza, la crescita e la risposta alle terapie dei tumori cerebrali.
Citazione: Manhrawy, I.I.M., Fathi, H., Alsekait, D.M. et al. Hybrid feature selection and classification model using high-dimensional data based on a metaheuristic algorithm for brain cancer diagnosis. Sci Rep 16, 11909 (2026). https://doi.org/10.1038/s41598-026-41573-5
Parole chiave: diagnosi del cancro cerebrale, espressione genica, selezione delle caratteristiche, apprendimento automatico, biomarcatori