Clear Sky Science · es
Selección de características híbrida y modelo de clasificación usando datos de alta dimensión basado en un algoritmo metaheurístico para el diagnóstico del cáncer cerebral
Por qué importan pruebas más inteligentes para los tumores cerebrales
Los tumores cerebrales están entre los cánceres más mortales, y sin embargo los médicos aún tienen dificultades para distinguir rápidamente y con precisión los distintos tipos de tumor. Las pruebas de laboratorio tradicionales pueden ser lentas, y las pruebas modernas basadas en genes generan un aluvión abrumador de datos: decenas de miles de mediciones génicas por paciente. Este estudio presenta un método informático que tamiza ese ruido genético para encontrar un pequeño conjunto de genes clave que ayudan a separar tumores peligrosos de casos menos agresivos, con el objetivo de respaldar un diagnóstico más preciso y, eventualmente, decisiones de tratamiento mejores.
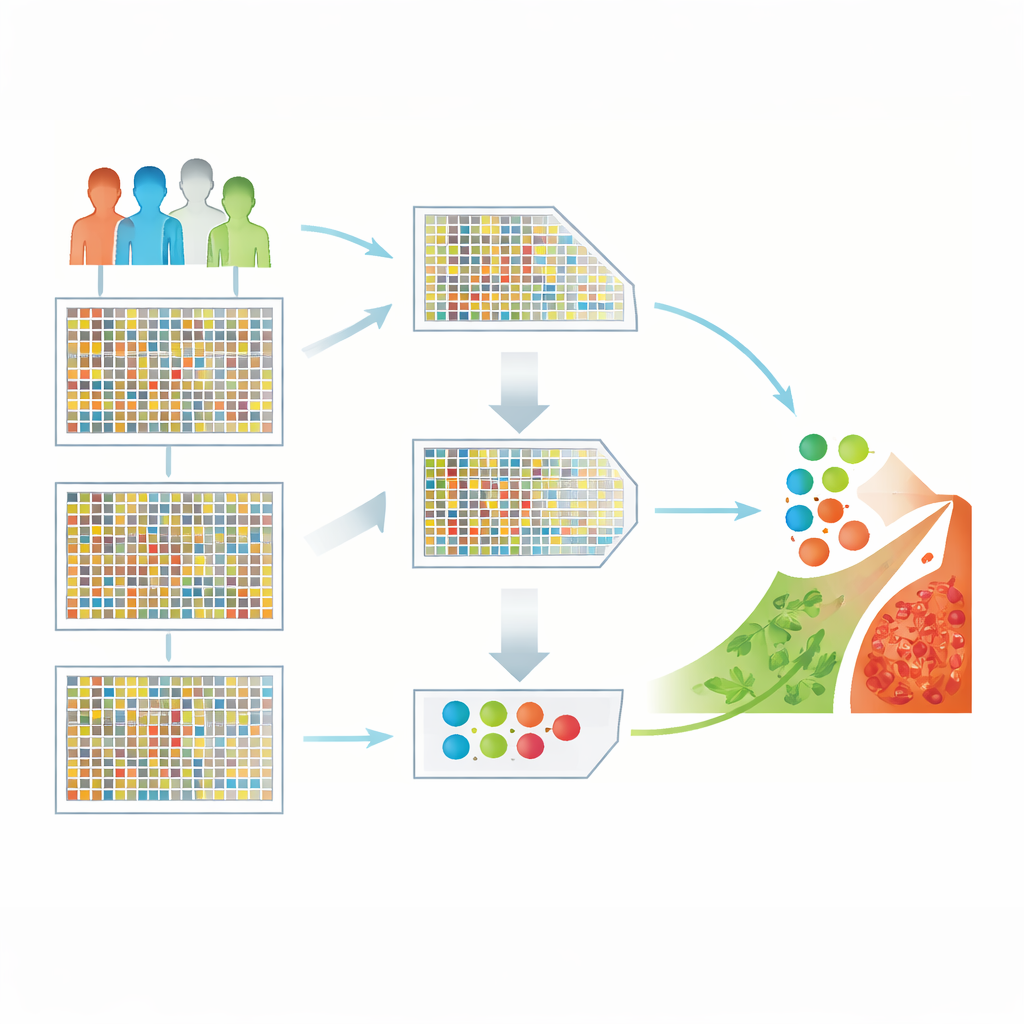
Convertir un mar de genes en pistas útiles
Los investigadores trabajaron con un conjunto de datos público de cáncer cerebral que contiene 130 muestras de pacientes, cada una medida en más de 54.000 genes. Datos de tan alta dimensión son poderosos pero complicados: hay muchas más mediciones génicas que pacientes, y muchos genes son irrelevantes o redundantes. Si se introducen directamente en un algoritmo de aprendizaje, este desequilibrio puede generar patrones engañosos y un rendimiento pobre en pacientes nuevos. El desafío central es conservar solo los genes más informativos mientras se descartan los demás, sin perder señales biológicas importantes.
Un enfoque en dos pasos para encontrar los genes más reveladores
Para abordar esto, el equipo diseñó una canalización híbrida de selección de características. En el primer paso, un filtro estadístico rápido llamado “redundancia mínima, relevancia máxima” (mRMR) analiza todos los genes y conserva aquellos que están fuertemente vinculados al cáncer cerebral y que, a la vez, son lo más distintos posible entre sí. Esto reduce rápidamente la lista de genes de decenas de miles a un conjunto más manejable. En el segundo paso, un método de búsqueda inspirado en el comportamiento de caza de aves rapaces—Harris Hawks Optimization—trata cada subconjunto posible de genes como una solución candidata y “caza” de forma iterativa combinaciones que ofrecen los mejores resultados de clasificación. Juntas, estas etapas reducen los 54.676 genes originales a solo 50 que aún capturan la esencia de la enfermedad.
Enseñar a las máquinas a trazar una línea diagnóstica más clara
Una vez identificado un conjunto compacto de genes, los autores entrenaron varios modelos de aprendizaje automático para distinguir entre cinco categorías de cáncer cerebral y, de forma más amplia, entre tejido maligno y menos peligroso. Un modelo clave, la máquina de vectores de soporte, necesita ajustar un par de parámetros de sensibilidad que afectan mucho su comportamiento. En lugar de afinarlos por prueba y error, el equipo utilizó tres estrategias de optimización—Optimización por Enjambre de Partículas, Evolución Diferencial y Harris Hawks Optimization—para buscar sistemáticamente los mejores ajustes. Evaluaron el rendimiento usando validación cruzada rigurosa y repetida y comprobaciones adicionales como remuestreo bootstrap y análisis de curvas de aprendizaje para asegurarse de que los modelos no estuvieran simplemente memorizando el conjunto de datos pequeño.
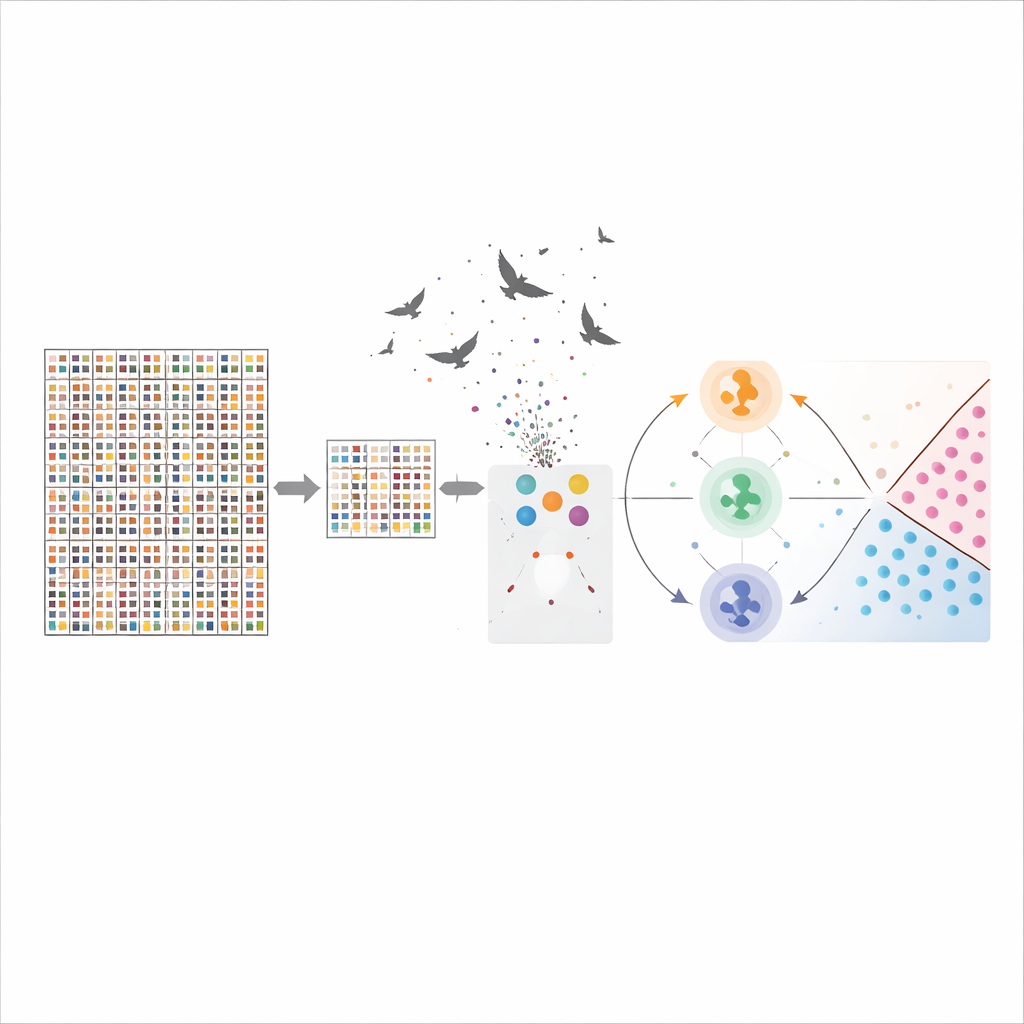
Qué tan bien funciona el sistema y qué significan los genes
Con la selección híbrida de genes y la optimización implementadas, la máquina de vectores de soporte alcanzó una precisión de alrededor del 96 % en los datos de cáncer cerebral, superando claramente a métodos más convencionales como los árboles de decisión. El método también redujo el número de genes de más de 54.000 a 50, preservando e incluso mejorando la capacidad predictiva. Muchos de los genes seleccionados ya se conocen por su implicación en el crecimiento celular, la reparación del ADN, la formación de vasos sanguíneos o la respuesta inmunitaria en tumores cerebrales, lo que da credibilidad biológica a las elecciones del ordenador. Esto significa que el modelo no solo es bueno para la clasificación, sino que también apunta a posibles biomarcadores que merecen ser estudiados en el laboratorio.
Qué podría significar esto para los pacientes
En términos sencillos, el estudio muestra que es posible comprimir un volumen enorme de datos genéticos en una pequeña “firma” génica significativa que ayuda a separar con alta fiabilidad distintos tipos de tumor cerebral. Aunque el trabajo todavía se basa en un grupo de pacientes relativamente pequeño y necesita ser probado en poblaciones más amplias y diversas, sugiere un camino hacia herramientas diagnósticas basadas en genes que sean rápidas, precisas e interpretables. Si se validan e integran en los flujos clínicos, dichas herramientas podrían proporcionar a los médicos evidencia más sólida al elegir tratamientos y ayudar a los científicos a centrarse en una lista corta de genes con más probabilidad de influir en cómo se inician, crecen y responden a la terapia los cánceres cerebrales.
Cita: Manhrawy, I.I.M., Fathi, H., Alsekait, D.M. et al. Hybrid feature selection and classification model using high-dimensional data based on a metaheuristic algorithm for brain cancer diagnosis. Sci Rep 16, 11909 (2026). https://doi.org/10.1038/s41598-026-41573-5
Palabras clave: diagnóstico de cáncer cerebral, expresión génica, selección de características, aprendizaje automático, biomarcadores