Clear Sky Science · it
VAP1 favorisce la fibrosi cardiaca abilitando il segnale PDGFR nei miofibroblasti
Perché la cicatrizzazione del cuore è importante
L’insufficienza cardiaca colpisce milioni di persone e spesso si sviluppa quando il cuore si riempie lentamente di tessuto cicatriziale rigido. Questo processo di cicatrizzazione, chiamato fibrosi, rende più difficile per il cuore rilassarsi e pompare sangue. Lo studio alla base di questo articolo esplora come certe cellule di supporto nel cuore si trasformano in cellule aggressive produttrici di cicatrici e identifica una molecola di superficie, VAP1, che favorisce questa transizione. Capire questo interruttore apre la strada a nuovi modi per rallentare o persino invertire la cicatrizzazione dannosa nelle malattie cardiache. 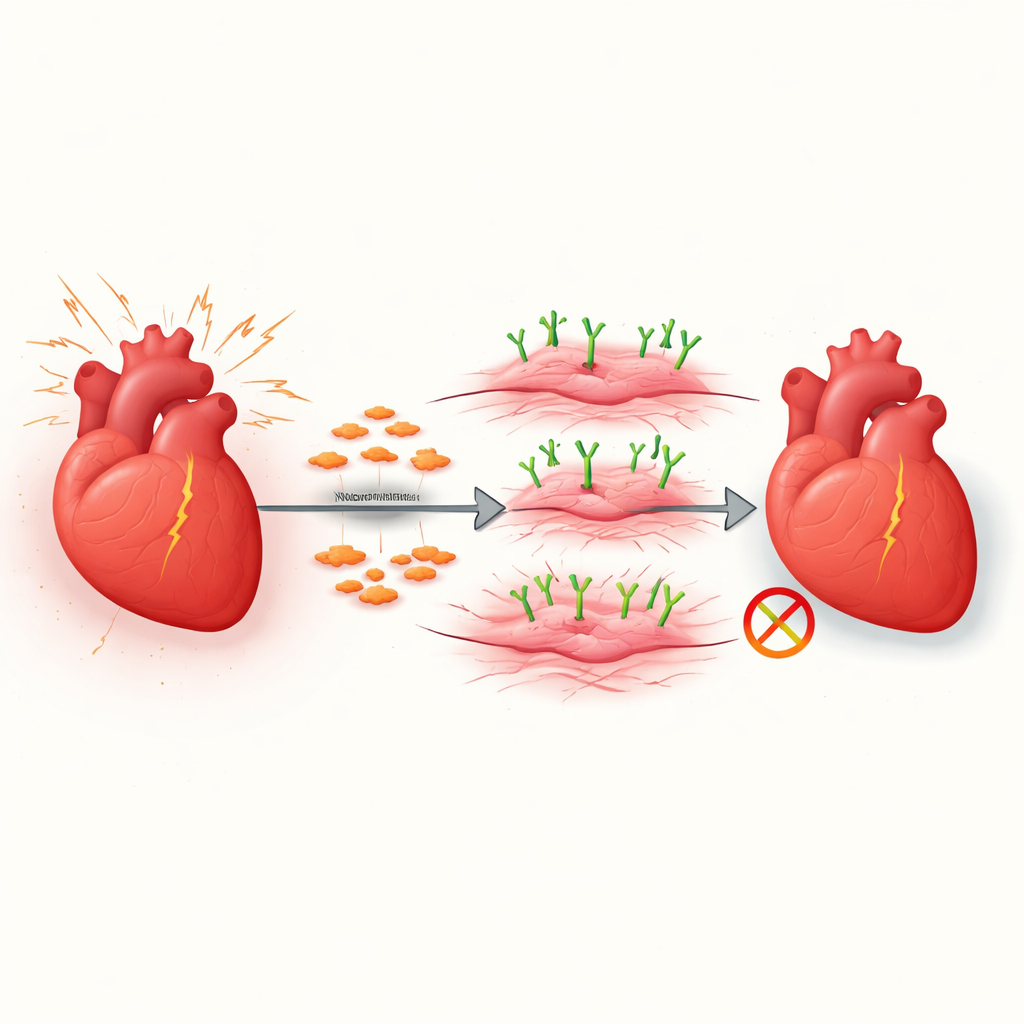
Da aiutanti silenziosi a costruttori iperattivi
Il cuore sano contiene molti fibroblasti, cellule di supporto silenziose che mantengono la struttura del tessuto. Quando il cuore è danneggiato o sottoposto a pressione a lungo termine, alcuni di questi fibroblasti si trasformano in miofibroblasti, che sono costruttori di tessuto cicatriziale altamente attivi. In brevi periodi questa risposta può essere protettiva, ma quando continua senza controllo riempie la parete cardiaca di fibre resistenti che ne limitano il movimento. I ricercatori si sono concentrati su come sia regolata questa transizione da fibroblasto a miofibroblasto, in particolare in un modello comune di sovraccarico di pressione che imita forme di insufficienza cardiaca umana.
Un guardiano di superficie chiamato VAP1
Utilizzando ampi screening di attività genica, il team ha scoperto che una proteina di superficie chiamata VAP1 aumentava nettamente quando veniva attivato un interruttore maestro della cicatrizzazione, la proteina MKL1. VAP1 è nota per aiutare i globuli bianchi ad aderire ai vasi sanguigni, ma il suo ruolo nel cuore era poco chiaro. In fibroblasti cardiaci coltivati in laboratorio, ridurre l’espressione di VAP1 attenuava caratteristiche tipiche dei miofibroblasti, come proliferazione cellulare, migrazione e capacità di contrarsi e tirare il loro ambiente. Aumentare VAP1 aveva l’effetto opposto, spingendo i fibroblasti più decisamente verso lo stato attivo e produttore di cicatrici. 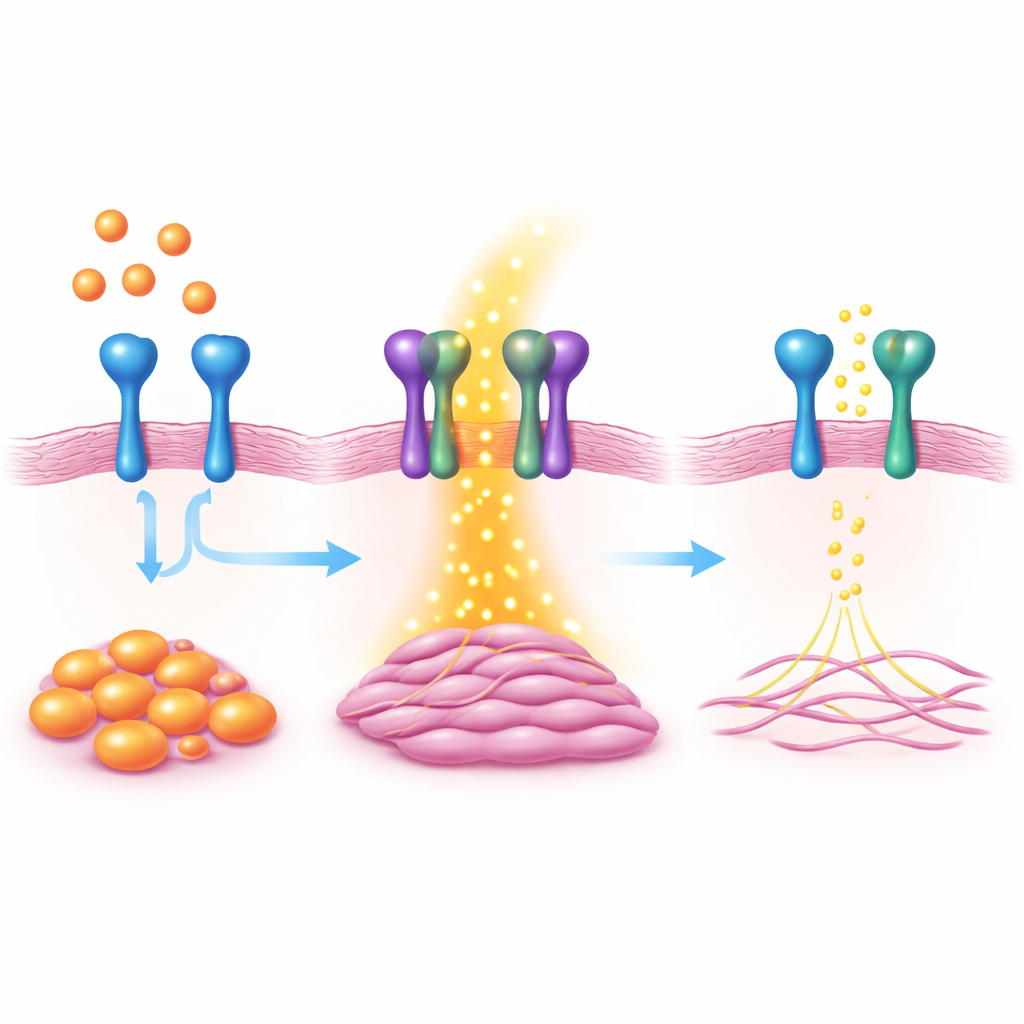
Testare VAP1 in cuori viventi
Per vedere come si comporta VAP1 in un organismo intero, gli scienziati hanno ingegnerizzato topi in cui VAP1 poteva essere rimosso solo dai fibroblasti cardiaci. Quando questi topi sono stati sottoposti a un sovraccarico di pressione prolungato sull’arteria principale che esce dal cuore, hanno sviluppato molta meno cicatrizzazione fibrosa rispetto ai topi normali, pur mostrando un ingrandimento cardiaco complessivo simile. Anche le misure della funzione di pompaggio cardiaco sono risultate meglio conservate. Rimuovere VAP1 specificamente dai miofibroblasti già attivati ha prodotto una riduzione simile della cicatrizzazione e un miglioramento della funzione, dimostrando che VAP1 è importante sia quando i fibroblasti si attivano sia mentre rimangono attivi.
Come VAP1 potenzia i segnali pro-cicatrice
Per scoprire come funziona VAP1, il team ha combinato l’analisi dell’attività genica con indagini proteiche. Hanno scoperto che VAP1 interagisce fisicamente con un’altra proteina di superficie chiamata PDGFRβ, un recettore che rileva segnali di crescita noti per innescare la fibrosi. Quando VAP1 mancava, PDGFRβ risultava meno attivo e una serie di vie di segnalazione a valle che promuovono proliferazione, migrazione cellulare e produzione di collagene venivano indebolite. Una porzione di VAP1 che si trova all’esterno della membrana cellulare è stata sufficiente a ripristinare questo segnale mancante quando è stata aggiunta di nuovo, suggerendo che la partnership fisica tra VAP1 e PDGFRβ sulla superficie cellulare è fondamentale.
Bloccare VAP1 con una piccola molecola
Poiché VAP1 funziona anche come enzima, i ricercatori hanno testato un composto di tipo farmacologico che ne inibisce l’attività. In fibroblasti cardiaci in coltura, questo inibitore ha ridotto i marcatori da miofibroblasto e limitato la loro capacità di dividersi, migrare e contrarsi. Somministrato a topi con sovraccarico di pressione, il composto non ha impedito l’ingrossamento cardiaco, ma ha ridotto sensibilmente l’accumulo di cicatrici e migliorato le misure della capacità di pompaggio del cuore. Questi risultati suggeriscono che il blocco farmacologico di VAP1 può attenuare la risposta di cicatrizzazione senza interrompere tutti gli aspetti dell’adattamento cardiaco allo stress.
Cosa significa per le persone con insufficienza cardiaca
Nel complesso, i risultati mostrano che VAP1 agisce come un coadiuvante cruciale per i segnali che spingono le cellule di supporto cardiache in uno stato rigido e produttore di cicatrici. Riducendo VAP1, sia geneticamente nei fibroblasti sia con un inibitore, il cuore sviluppa meno collagene rigido e mantiene una funzione migliore sotto stress. Pur richiedendo ulteriori studi su altri modelli animali e su tessuto umano, lo studio pone VAP1 come un promettente bersaglio per future terapie mirate a limitare la fibrosi cardiaca dannosa e ad alleviare il carico dell’insufficienza cardiaca.
Citazione: Huang, S., Zhao, Q., Shao, T. et al. VAP1 promotes cardiac fibrosis by enabling PDGFR signaling in myofibroblasts. Exp Mol Med 58, 1284–1296 (2026). https://doi.org/10.1038/s12276-026-01690-7
Parole chiave: fibrosi cardiaca, insufficienza cardiaca, fibroblasti, VAP1, segnalazione PDGFR