Clear Sky Science · fr
Un prédicteur pharmacocinétique basé sur l’apprentissage automatique (EGFR-PROPK) pour les PROTACs ciblant l’EGFR
Conception de médicaments plus intelligente pour des cancers difficiles à traiter
Beaucoup de nouveaux médicaments anticancéreux prometteurs échouent non pas parce qu’ils manquent leur cible, mais parce que l’organisme les élimine trop rapidement, les stocke au mauvais endroit ou les absorbe mal. Une nouvelle classe de médicaments appelée PROTACs peut détruire des protéines causant la maladie qui étaient jadis considérées comme « non ciblables », mais leur comportement dans l’organisme est particulièrement difficile à prévoir. Cette étude présente un outil d’apprentissage automatique, EGFR-PROPK, conçu spécifiquement pour prédire comment un groupe de PROTACs dirigés contre une protéine clé du cancer se déplacent dans l’organisme, dans le but d’aider les chercheurs à choisir plus tôt de meilleurs candidats-médicaments et à éviter de perdre des années et des ressources sur des impasses.
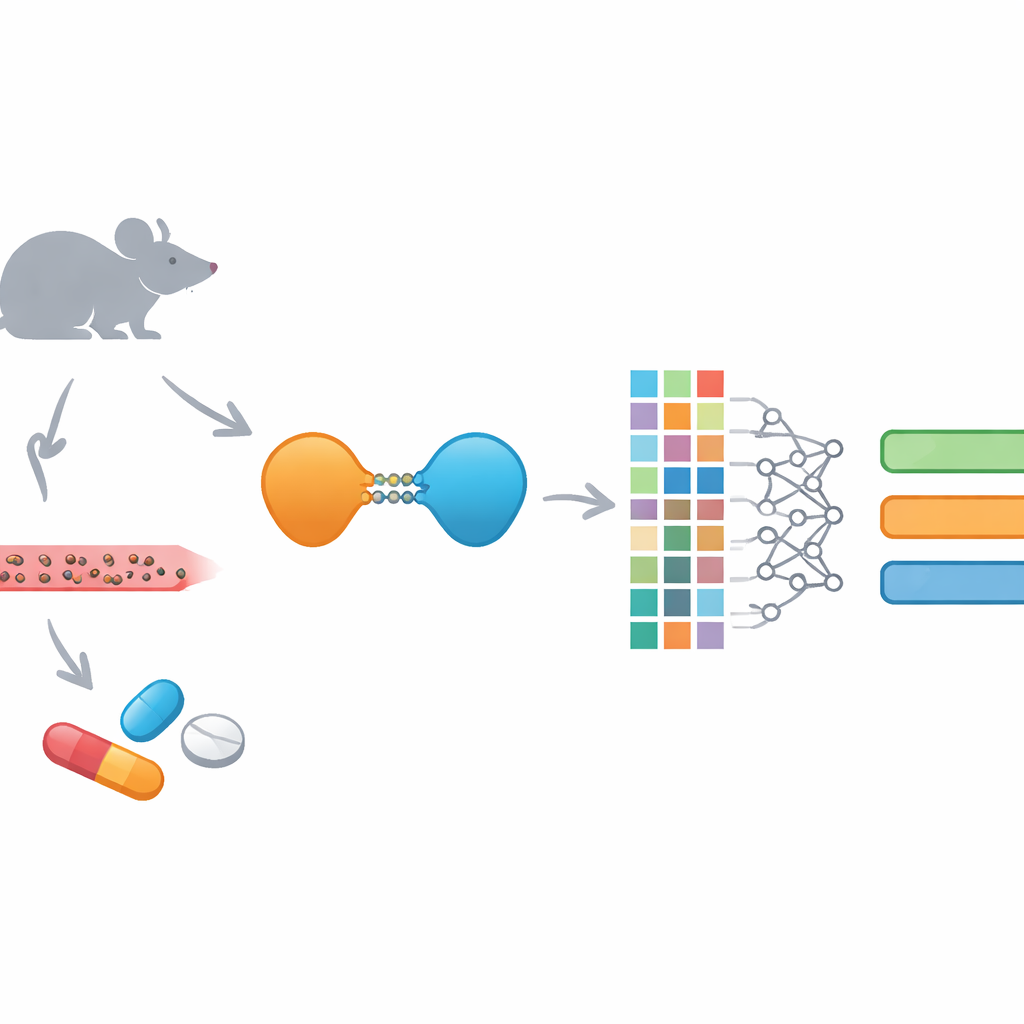
Nouveaux médicaments destructeurs de protéines et leurs défis
Les PROTACs sont des médicaments inhabituels : au lieu de simplement bloquer une protéine, ils la marquent pour destruction par la machinerie d’élimination de la cellule. Chaque PROTAC relie un élément de liaison pour la cible pathologique à un autre élément qui recrute un « destructeur » cellulaire, reliés par une chaîne flexible. Cette conception leur permet d’atteindre des protéines que les médicaments traditionnels ne peuvent pas facilement cibler et autorise souvent une liaison temporaire plutôt qu’une occupation constante du site protéique. Cependant, cette même taille et complexité rendent les PROTACs difficiles à délivrer et à contrôler dans l’organisme. Ils ont tendance à être volumineux, relativement lipophiles et fortement liés aux protéines plasmatiques, autant de facteurs qui compliquent leur absorption, distribution, métabolisme et élimination.
Pourquoi les règles habituelles du comportement des médicaments sont insuffisantes
Les développeurs traditionnels de médicaments utilisent des nombres simples, tels que le poids moléculaire, l’affinité pour les graisses (LogP) et des règles empiriques de « drug-likeness », pour estimer le comportement d’un composé dans l’organisme. Les chercheurs ont commencé par se demander si ces mesures familières pouvaient expliquer comment 100 PROTACs ciblant le récepteur du facteur de croissance épidermique (EGFR) — un moteur majeur de nombreux cancers — se comportent chez la souris vivante. Ils se sont concentrés sur trois propriétés clés : la clairance (vitesse d’élimination par l’organisme), la demi-vie (durée de présence en circulation) et le volume de distribution apparent (amplitude de diffusion dans les tissus). Lorsqu’ils ont comparé ces propriétés à des descripteurs basiques, les points de données étaient dispersés sans tendances claires, montrant que des règles chimiques simples ne suffisent pas à prédire le comportement des PROTACs.
Entraîner un moteur de prédiction sur mesure
Pour combler cette lacune, l’équipe a construit EGFR-PROPK, un prédicteur d’apprentissage automatique spécifiquement ajusté aux PROTACs dirigés contre l’EGFR. Ils ont d’abord réalisé des études chez la souris sur 100 PROTACs différents, mesurant leur clairance, demi-vie et volume de distribution après administration. Ils ont ensuite décrit chaque molécule à l’aide de plusieurs « empreintes » détaillées codant sa structure et calculé 200 caractéristiques chimiques supplémentaires. Ces descriptions numériques ont été introduites dans un modèle basé sur CatBoost, un algorithme moderne performant pour détecter des motifs dans des données tabulaires. À la différence des outils polyvalents, EGFR-PROPK a été entraîné directement sur des résultats in vivo pour des PROTACs, ce qui lui permet d’apprendre des relations subtiles entre la forme complexe des linkers, la taille globale et la durée de présence des médicaments dans le sang ou leur diffusion dans les tissus.
Surpasser les modèles généraux et révéler des nuances cachées
Les chercheurs ont comparé EGFR-PROPK à des plateformes de prédiction largement utilisées, principalement conçues pour des petites molécules traditionnelles. Lorsque ces modèles génériques ont été appliqués « tels quels » aux PROTACs, leurs prédictions étaient médiocres : les valeurs estimées de demi-vie, de clairance et de distribution tissulaire déviaient souvent fortement des mesures réelles. Même après qu’un modèle concurrent ait été ajusté avec des données PROTAC, il restait en deçà des performances. En revanche, EGFR-PROPK a montré une concordance nettement meilleure entre valeurs prédites et expérimentales, en particulier pour la demi-vie et la clairance, avec des gains modérés pour la distribution. Le modèle a également géré des cas réels : deux PROTACs de structures très similaires mais de comportements très différents ont été distingués plus précisément par EGFR-PROPK que par les outils généraux, soulignant l’importance de données d’entraînement spécifiques aux PROTACs.
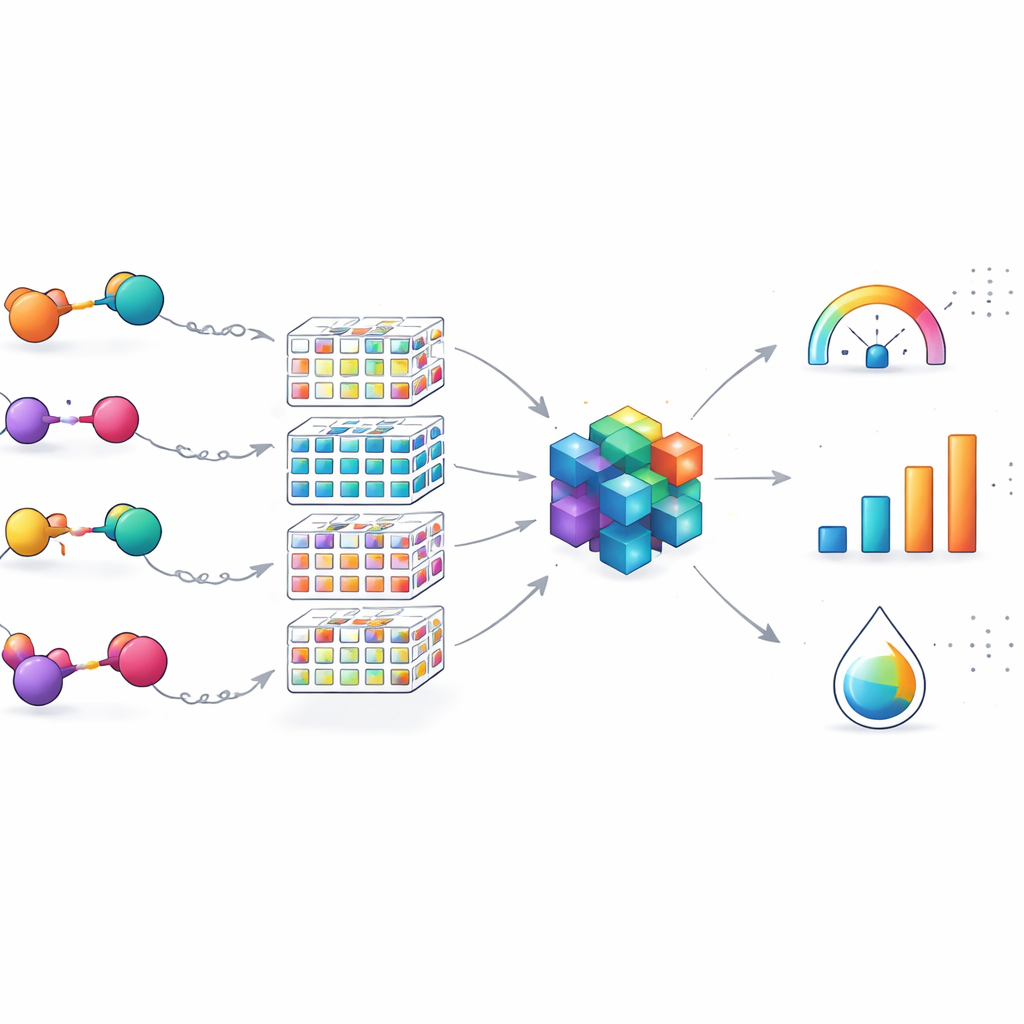
Cartographier le paysage chimique et ses limites
Au-delà des chiffres de performance, l’équipe a vérifié si leurs 100 PROTACs couvraient une large gamme de conceptions ou seulement un coin étroit de l’espace chimique. En réduisant les molécules à leurs échafaudages de base et en les visualisant parmi des milliers de PROTACs brevetés, ils ont montré que leur ensemble couvre de nombreux types structurels distincts, en particulier pour les linkers flexibles. Cela signifie que le modèle convient bien pour affiner des PROTACs dirigés contre l’EGFR qui partagent des éléments de base mais diffèrent par la façon dont leurs parties sont reliées. Dans le même temps, les auteurs insistent sur le fait que l’outil est un spécialiste de domaine : sa précision peut décliner pour des PROTACs construits autour de cibles ou de voies de dégradation entièrement différentes, ce qui souligne la nécessité d’une extension future et d’un apprentissage par transfert entre jeux de données.
Ce que cela signifie pour les médicaments de demain
Pour les non-spécialistes, l’idée principale est que la « plomberie » d’un médicament dans l’organisme — comment il est absorbé, où il se rend et combien de temps il y reste — peut faire ou défaire une thérapie anticancéreuse prometteuse, et que les raccourcis habituels pour les petites pilules ne fonctionnent pas bien pour des dégradeurs de nouvelle génération comme les PROTACs. En créant EGFR-PROPK, les chercheurs montrent que des données animales soigneusement collectées, combinées à des méthodes avancées de reconnaissance de motifs, peuvent fournir une vision beaucoup plus précise dès les premières étapes de la façon dont ces molécules complexes se comporteront. Ce type de prédiction sur mesure devrait aider les chimistes à concevoir des PROTACs qui non seulement détruisent les bonnes protéines mais les atteignent aussi aux bons niveaux, ouvrant la voie à des traitements plus efficaces et plus sûrs contre les cancers résistants.
Citation: Zhang, R., Li, F., Liu, Y. et al. A machine learning-based pharmacokinetics predictor (EGFR-PROPK) for EGFR-targeting PROTACs. Commun Chem 9, 134 (2026). https://doi.org/10.1038/s42004-026-01938-3
Mots-clés: Pharmacocinétique des PROTACs, Dégradeurs ciblant l’EGFR, Apprentissage automatique en conception de médicaments, Prédiction ADMET, Dégradation ciblée des protéines