Clear Sky Science · en
A machine learning-based pharmacokinetics predictor (EGFR-PROPK) for EGFR-targeting PROTACs
Smarter Drug Design for Tough-to-Treat Cancers
Many promising new cancer drugs fail not because they miss their target, but because the body clears them too quickly, stores them in the wrong places, or cannot absorb them well. A new class of medicines called PROTACs can destroy disease-causing proteins that were once considered “undruggable,” but their behavior inside the body is especially hard to predict. This study introduces a machine learning tool, EGFR-PROPK, designed specifically to forecast how a group of PROTACs against a key cancer protein move through the body, with the goal of helping scientists pick better drug candidates earlier and waste fewer years and resources on dead ends.
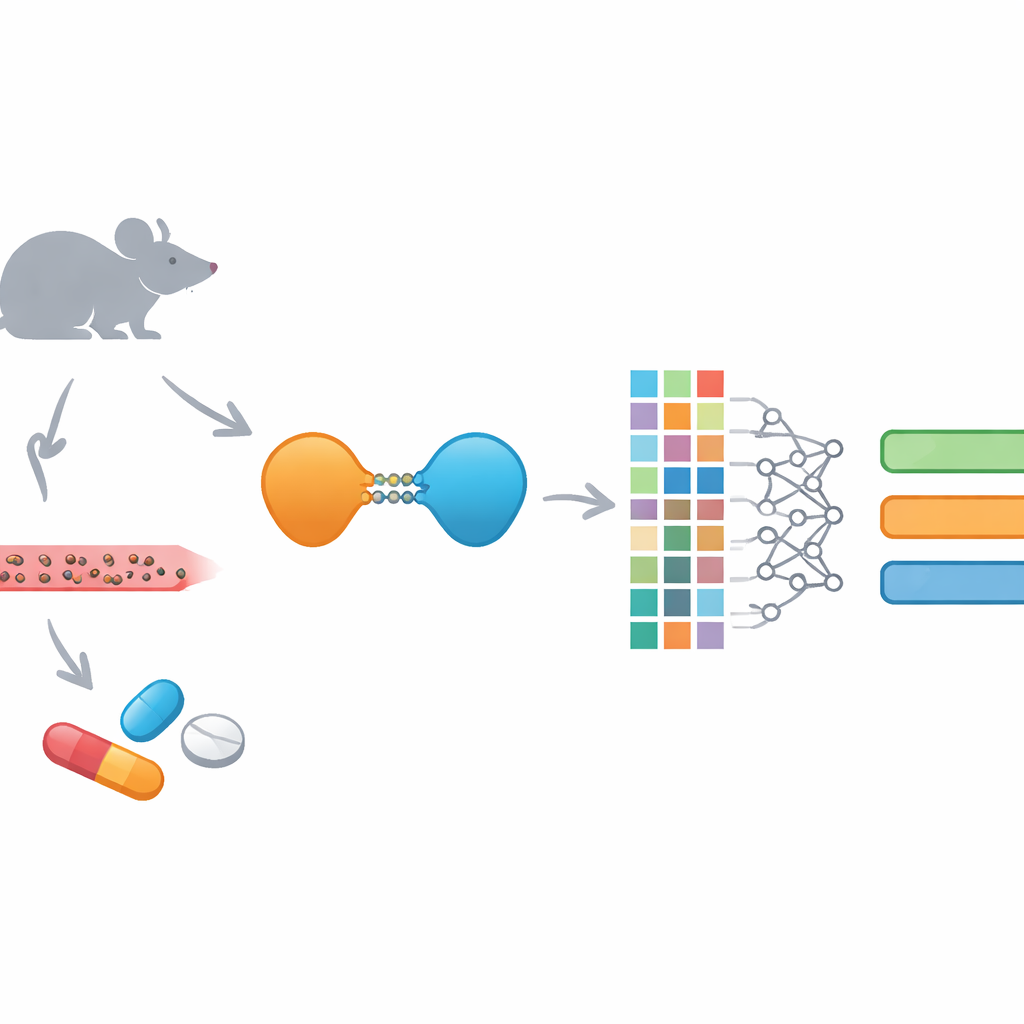
New Protein-Destroying Drugs and Their Challenges
PROTACs are unusual drugs: instead of just blocking a protein, they tag it for destruction by the cell’s own disposal machinery. Each PROTAC links a binding piece for the disease target to another piece that recruits a cellular “shredder,” joined by a flexible chain. This design lets them go after proteins that normal drugs cannot easily touch and often allows temporary binding rather than constant occupation of a protein site. However, that same size and complexity make PROTACs difficult to deliver and control inside the body. They tend to be large, relatively greasy, and strongly bound to blood proteins, all of which complicate how they are absorbed, distributed, broken down, and removed.
Why Usual Rules for Drug Behavior Fall Short
Traditional drug developers use simple numbers, such as molecular weight, fat-liking (LogP), and rule-of-thumb measures of “drug-likeness,” to guess how a compound might behave in the body. The researchers began by asking whether these familiar measures could explain how 100 PROTACs that target the epidermal growth factor receptor (EGFR)—a major driver in many cancers—behave in live mice. They focused on three key properties: clearance (how fast the body removes the drug), half-life (how long the drug stays in circulation), and apparent volume of distribution (how widely it spreads into tissues). When they compared these properties to basic descriptors, the data points were scattered with almost no clear trends, showing that simple chemical rules are not enough to predict how PROTACs behave.
Training a Tailor-Made Prediction Engine
To overcome this gap, the team built EGFR-PROPK, a machine learning predictor tuned specifically to EGFR-targeting PROTACs. They first carried out mouse studies on 100 different PROTACs, measuring their clearance, half-life, and volume of distribution after dosing. They then described each molecule using several rich “fingerprints” that encode its detailed structure and calculated 200 additional chemical features. These numerical descriptions were fed into a CatBoost-based model, a modern algorithm that excels at finding patterns in tabular data. Unlike general-purpose tools, EGFR-PROPK was trained directly on in-vivo results for PROTACs, allowing it to learn subtle relationships between complex linker shapes, overall size, and how long the drugs remain in blood or spread into tissues.
Outperforming General Models and Revealing Hidden Nuances
The researchers compared EGFR-PROPK with widely used prediction platforms built mostly for traditional small-molecule drugs. When those generic models were applied “as is” to PROTACs, their predictions were poor: estimated values for half-life, clearance, and tissue distribution often deviated strongly from real measurements. Even after one competitor model was fine-tuned with PROTAC data, it still lagged behind. In contrast, EGFR-PROPK achieved substantially stronger agreement between predicted and experimental values, especially for half-life and clearance, with moderate gains for distribution. The model also handled real-world case studies: two PROTACs with very similar structures but very different behavior were more accurately distinguished by EGFR-PROPK than by general tools, underscoring the importance of PROTAC-specific training data.
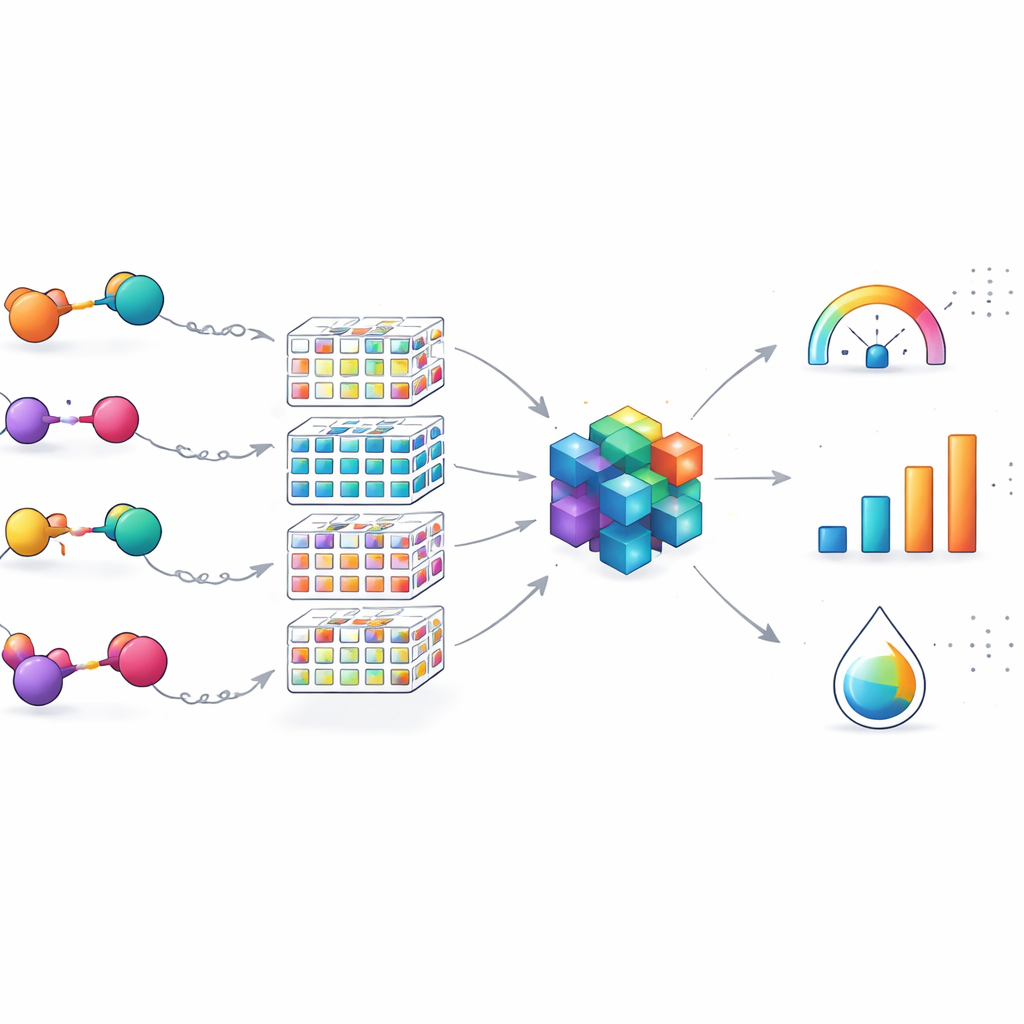
Mapping the Chemical Landscape and Its Limits
Beyond performance numbers, the team checked whether their 100 PROTACs covered a broad range of designs or just a narrow corner of chemical space. By stripping molecules down to their core scaffolds and visualizing them among thousands of patented PROTACs, they showed that their set spans many distinct structural types, especially in the flexible linkers. This means the model is well suited for refining EGFR-directed PROTACs that share key building blocks but vary in how their parts are connected. At the same time, the authors stress that the tool is a domain specialist: its accuracy may drop for PROTACs built around entirely different targets or degradation pathways, pointing to the need for future expansion and transfer learning across datasets.
What This Means for Future Medicines
For non-experts, the main takeaway is that the “plumbing” of a drug in the body—how it is absorbed, where it goes, and how long it stays—can make or break a promising cancer therapy, and the usual shortcuts for small pills do not work well for next-generation degraders like PROTACs. By creating EGFR-PROPK, the researchers show that carefully collected animal data, combined with advanced pattern-recognition methods, can provide a much sharper early look at how these complex molecules will behave. This kind of tailored prediction should help chemists design PROTACs that not only destroy the right proteins but also reach them at the right levels, paving the way for more effective and safer treatments against resistant cancers.
Citation: Zhang, R., Li, F., Liu, Y. et al. A machine learning-based pharmacokinetics predictor (EGFR-PROPK) for EGFR-targeting PROTACs. Commun Chem 9, 134 (2026). https://doi.org/10.1038/s42004-026-01938-3
Keywords: PROTAC pharmacokinetics, EGFR-targeted degraders, machine learning in drug design, ADMET prediction, targeted protein degradation