Clear Sky Science · fr
Apprentissage automatique interprétable pour la conception de matériaux thermoélectriques avec les réseaux de Kolmogorov–Arnold
Transformer la chaleur en énergie utile
Chaque jour, d’énormes quantités d’énergie sont perdues sous forme de chaleur résiduelle provenant des moteurs de voiture, des usines et même de nos appareils électroniques. Les matériaux thermoélectriques offrent un moyen de convertir une partie de cette chaleur perdue directement en électricité, sans pièces mobiles et sans carburant. Mais découvrir de nouveaux matériaux efficaces est difficile, car leurs performances dépendent de plusieurs propriétés électroniques étroitement liées. Cette étude explore un nouveau type d’intelligence artificielle qui peut non seulement prédire la qualité d’un matériau, mais aussi expliquer pourquoi — ouvrant une voie plus claire pour concevoir de meilleurs composés thermoélectriques.
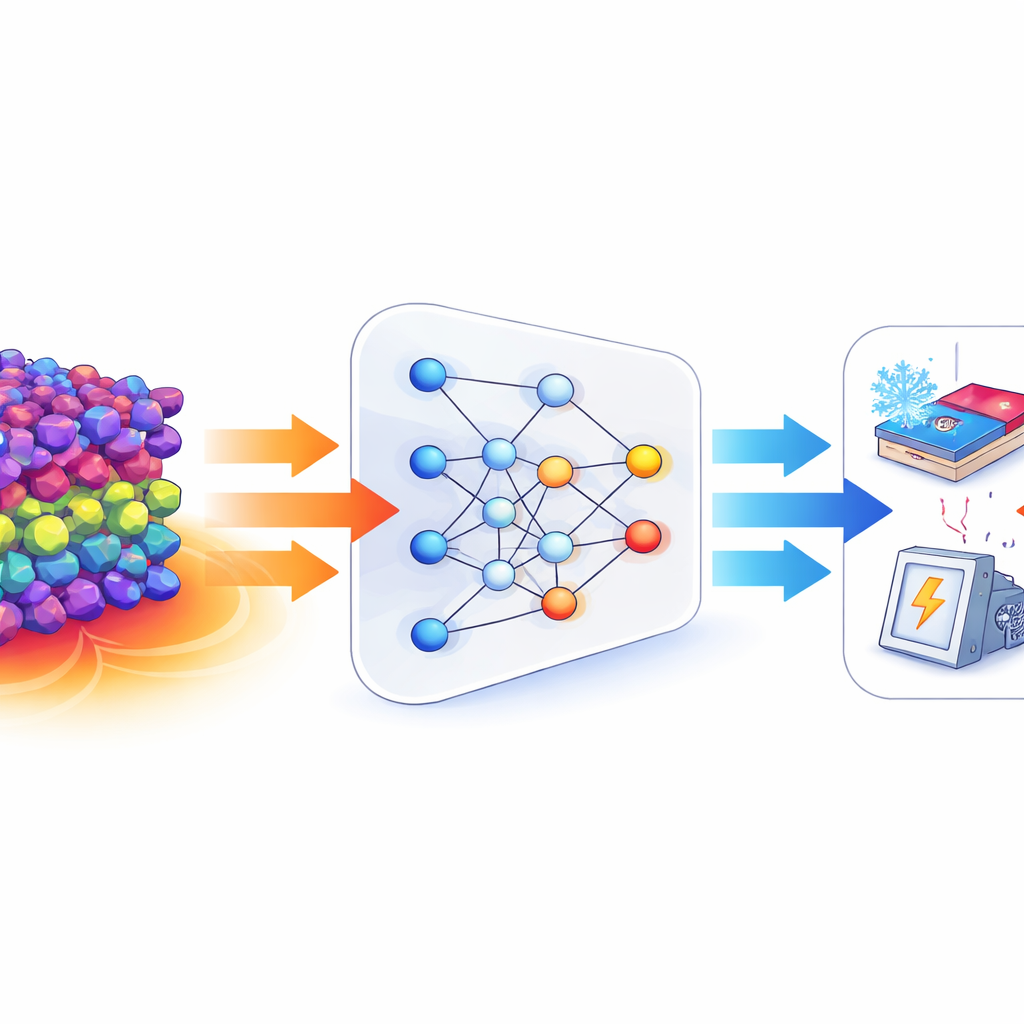
Pourquoi il est difficile de trouver de meilleurs matériaux de conversion chaleur-électricité
Les dispositifs thermoélectriques reposent sur des matériaux capables de générer une tension lorsqu’un côté est chaud et l’autre est froid. Leur efficacité est résumée par une grandeur appelée zT, qui dépend de trois ingrédients principaux : la sensibilité des porteurs de charge à la température (le coefficient de Seebeck), la facilité avec laquelle ils se déplacent (la conductivité électrique) et la capacité du matériau à conduire la chaleur (la conductivité thermique). Améliorer l’un de ces termes nuit souvent à un autre — par exemple, augmenter la conductivité électrique peut aussi augmenter la conductivité thermique, ce qui réduit l’efficacité. Les simulations quantiques traditionnelles, comme la théorie de la fonctionnelle de la densité, peuvent prédire ces propriétés à partir de la structure atomique, mais elles sont si coûteuses en calcul qu’elles ne peuvent pas être appliquées à des millions de matériaux candidats.
Modèles boîte noire et besoin d’explications
Les modèles d’apprentissage automatique sont récemment devenus des outils puissants pour estimer rapidement les propriétés des matériaux à partir de simulations et d’expériences passées. Dans ce travail, les auteurs partent de descriptions numériques riches des structures cristallines produites par un encodeur spécialisé « Crystalformer », qui capture la façon dont les atomes sont disposés et interagissent dans un cristal. Ils entraînent d’abord un perceptron multicouche standard — un modèle de deep learning courant — pour prédire deux quantités clés : le coefficient de Seebeck et la bande interdite électronique, qui influence la facilité avec laquelle un matériau peut héberger des porteurs de charge mobiles. Ce modèle atteint une grande précision sur les deux tâches sur un vaste jeu de données d’environ 15 000 composés. Cependant, comme la plupart des réseaux profonds, il se comporte comme une boîte noire, offrant peu d’indications sur les caractéristiques structurales réellement importantes ou sur la façon dont elles se combinent pour contrôler la réponse thermoélectrique.
Un type différent de réseau neuronal
L’idée centrale de l’article est de remplacer les réseaux neuronaux opaques par des réseaux de Kolmogorov–Arnold, ou KAN. Plutôt que de dissimuler toute la complexité dans les activations neuronales, les KAN attachent des fonctions flexibles et unidimensionnelles, semblables à des courbes, aux connexions entre couches. Mathématiquement, ces courbes sont des splines lisses qui s’adaptent aux données, et le modèle global peut s’écrire comme une somme de fonctions simples des descripteurs en entrée. Après l’apprentissage, les auteurs ajustent des expressions mathématiques concises — construites à partir de fonctions familières comme les sinus, cosinus et courbes de saturation lisses — à ces splines apprises. De cette façon, le modèle devient une formule symbolique explicite reliant les descripteurs structurels à la bande interdite et au coefficient de Seebeck, plutôt qu’un enchevêtrement de paramètres opaques. Bien que les KAN soient plus coûteux à entraîner, ils atteignent une précision comparable, et dans certains régimes supérieure, à celle du perceptron multicouche et à plusieurs autres références d’apprentissage automatique de la littérature.
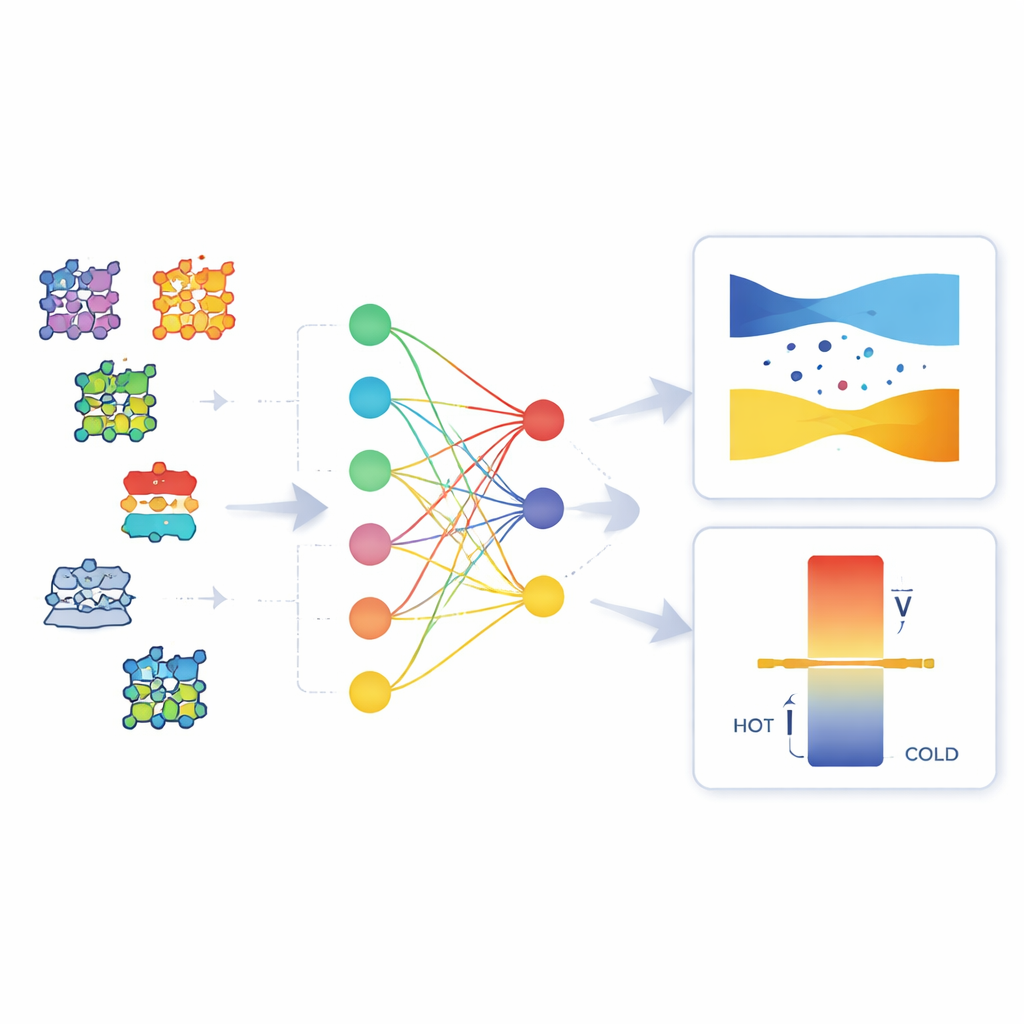
Ce que le modèle apprend sur les matériaux
En examinant la structure interne du KAN, les auteurs montrent qu’un seul sous-ensemble des 128 descripteurs structurels influence fortement chaque propriété. Ils calculent des scores d’attribution pour identifier les descripteurs les plus importants, puis élaguent les connexions faibles, obtenant un réseau épars, plus facile à interpréter, qui prédit toujours bien. En utilisant des paires de descripteurs les mieux classés, ils construisent des cartes bidimensionnelles qui montrent comment la bande interdite prédite ou le coefficient de Seebeck varie dans l’espace des descripteurs. Ces cartes révèlent des effets lisses et coopératifs, où des tendances oscillatoires et des saturations se combinent, plutôt que des règles simples un à un. D’un point de vue chimique, le modèle peine davantage dans les systèmes complexes à liaisons fortement directionnelles ou avec des électrons d et f fortement corrélés, mais même là les KAN fournissent des prédictions plus stables et physiquement sensées que le modèle boîte noire. Par exemple, ils produisent moins de bandes interdites négatives non physiques et moins de valeurs de Seebeck au signe erroné.
De la prédiction à la conception guidée
Parce que les KAN donnent en sortie des formules explicites et lisses plutôt que de simples prédictions numériques, ils peuvent être utilisés pour explorer comment la modification des descripteurs sous-jacents ferait évoluer la performance thermoélectrique. Bien que ces descripteurs restent des quantités abstraites dérivées d’un encodeur structurel profond, ils corrèlent avec des caractéristiques réelles des arrangements atomiques et des liaisons. Cela signifie que les chercheurs peuvent utiliser le modèle analytique KAN comme une sorte de carte : en le sondant pour repérer des régions associées à un coefficient de Seebeck élevé et à une bande interdite adaptée, ils peuvent réduire la liste de matériaux prometteurs, puis relier ces motifs de descripteurs à des structures cristallines candidates dans de grandes bases de données ou des outils de conception générative.
Ce que cela signifie pour les matériaux futurs
Pour un lecteur non spécialiste, le message clé est que ce travail nous rapproche d’une intelligence artificielle « boîte de verre » pour la découverte de matériaux. Les réseaux de Kolmogorov–Arnold offrent des performances proches de celles des meilleurs prédicteurs boîte noire, mais ils exposent aussi, sous forme mathématique, la façon dont les caractéristiques structurelles d’un cristal sont liées à sa capacité à convertir la chaleur en électricité. Cette combinaison de rapidité, de précision et d’interprétabilité peut aider les scientifiques à faire des choix éclairés sur les nouveaux composés à synthétiser et tester, raccourcissant potentiellement le chemin entre les données brutes et des dispositifs thermoélectriques réels qui récupèrent la chaleur perdue et améliorent l’efficacité énergétique.
Citation: Fronzi, M., Ford, M.J., Nayal, K.S. et al. Interpretable machine learning for thermoelectric materials design with Kolmogorov–Arnold networks. Sci Rep 16, 14146 (2026). https://doi.org/10.1038/s41598-026-44723-x
Mots-clés: matériaux thermoélectriques, apprentissage automatique interprétable, réseaux de Kolmogorov–Arnold, conception de matériaux, coefficient de Seebeck