Clear Sky Science · es
La quinasa proteica DYRK1B es un gen diana de p53 y funciona como un regulador de retroalimentación negativo del factor de transcripción RFX7
Por qué esto importa para el tratamiento del cáncer
La mayoría de los tumores presentan daño en el famoso “guardián del genoma”, una proteína llamada p53. Este guardián normalmente ayuda a las células a detenerse, reparar su ADN o autodestruirse cuando algo va mal. El estudio resumido aquí descubre una nueva forma en que las células cancerosas pueden debilitar este sistema protector a través de otra proteína, una quinasa llamada DYRK1B, y muestra que bloquear DYRK1B podría hacer que las células tumorales sean más vulnerables a la quimioterapia.
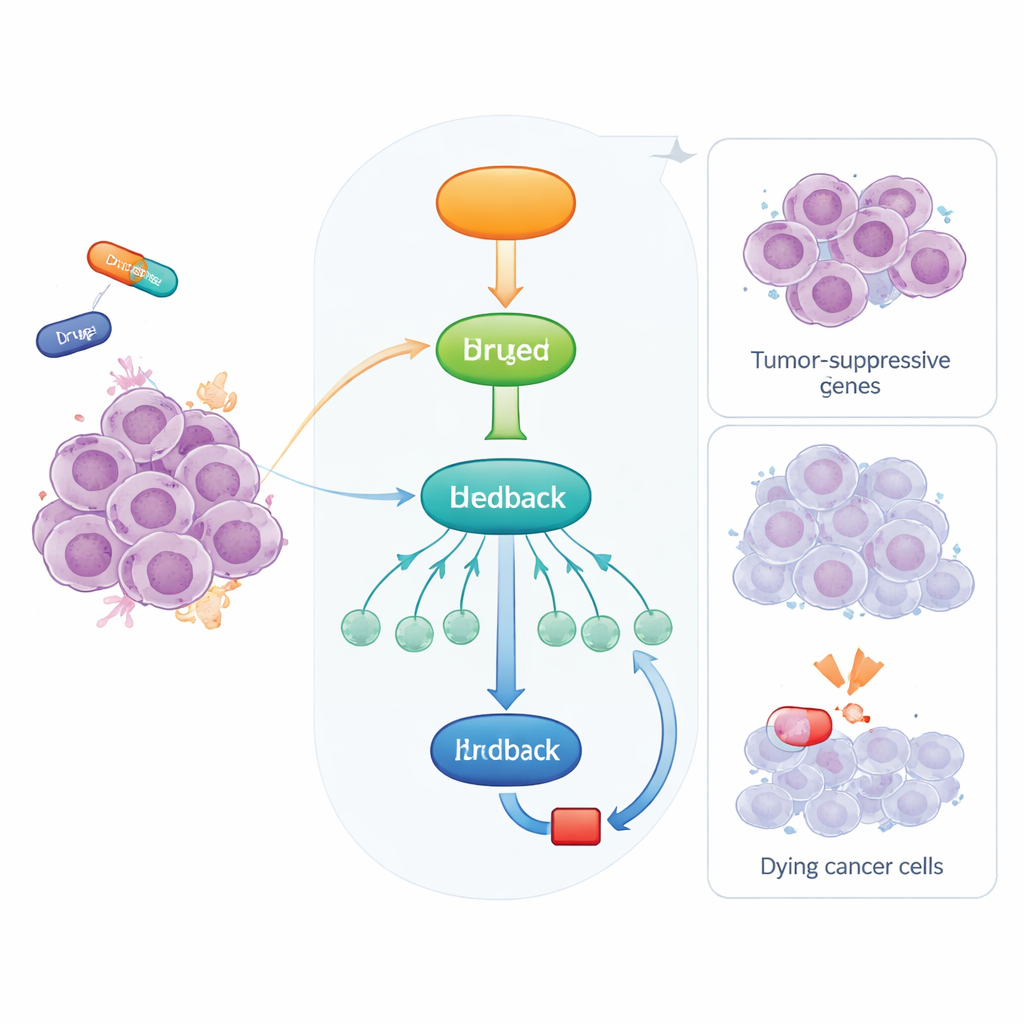
Un sistema de alarma incorporado en nuestras células
Cuando las células experimentan estrés, como daño en el ADN o problemas al sintetizar componentes celulares, p53 se activa y lanza un programa de emergencia. Lo hace en gran parte regulando la expresión de otros genes. Algunos de esos genes detienen directamente el ciclo celular o desencadenan la muerte celular, pero p53 también actúa a través de proteínas de control adicionales que regulan a su vez numerosos genes aguas abajo. Una de ellas es RFX7, un factor de transcripción que ha emergido recientemente como un importante supresor tumoral. RFX7 ayuda a activar una red de genes que restringen el crecimiento tumoral y con frecuencia está alterado o silenciado en cánceres humanos.
Una quinasa que favorece la supervivencia en el foco
DYRK1B es una enzima que añade grupos fosfato a otras proteínas, cambiando así su comportamiento. Trabajos previos mostraron que DYRK1B ayuda a las células cancerosas a soportar condiciones adversas, mantener un estado de baja actividad y reparar el daño en el ADN. Se encuentra con frecuencia en niveles anormalmente altos en varios tumores sólidos, y bloquearla puede aumentar la sensibilidad de las células cancerosas a la quimioterapia o a la radiación en modelos experimentales. Sin embargo, en comparación con muchas otras enzimas relacionadas con el cáncer, DYRK1B ha permanecido poco comprendida, ganando la etiqueta de “quinasa oscura”. El nuevo estudio se propuso aclarar cómo se controla DYRK1B y cómo encaja en el circuito más amplio de respuesta al estrés gobernado por p53.
De p53 a RFX7 y luego a DYRK1B
Los investigadores trataron diversas líneas celulares cancerosas con dos fármacos quimioterapéuticos, doxorrubicina y actinomicina D, ambos activadores de p53. Observaron que los niveles de DYRK1B aumentaban notablemente tras el tratamiento, mientras que su par cercano DYRK1A no lo hacía. Usando un fármaco llamado Nutlin-3a, que activa p53 sin causar daño en el ADN, confirmaron que DYRK1B se induce siempre que p53 se activa. Cuando p53 fue eliminado genéticamente, este aumento de DYRK1B desapareció, y el análisis de datos tumorales de pacientes mostró que la expresión de DYRK1B tiende a correlacionar con los niveles de p53 en muchos tipos de cáncer. El equipo mostró además que esta inducción es indirecta: p53 estimula primero a RFX7, y RFX7 a su vez impulsa a DYRK1B. La eliminación de RFX7 o la inhabilitación de su capacidad para entrar en el núcleo redujeron drásticamente la inducción de DYRK1B, y los niveles de DYRK1B aumentaron tanto a nivel de ARN como de proteína, confirmando una activación génica genuina.
Un freno molecular sobre un supresor tumoral
Una vez producido, DYRK1B no permanece inactivo. El estudio revela que DYRK1B se asocia físicamente con RFX7 en las células y lo modifica. Cuando p53 activa a RFX7, la proteína adopta una forma asociada a una fuerte actividad activadora de genes. Inhibir DYRK1B con pequeñas moléculas o reducirla mediante un degradador dirigido potencia esta forma activa de RFX7, mientras que sobreexpresar DYRK1B convierte a RFX7 de nuevo en un estado menos activo y atenúa la producción de varias proteínas supresoras tumorales controladas por RFX7, incluyendo PDCD4. Experimentos bioquímicos muestran que DYRK1B fosforila la región terminal de RFX7, provocando que migre de manera diferente en geles y pierda potencia transcripcional. En esencia, DYRK1B forma un circuito de retroalimentación negativo: p53 activa RFX7, RFX7 eleva DYRK1B, y DYRK1B a su vez frena a RFX7.
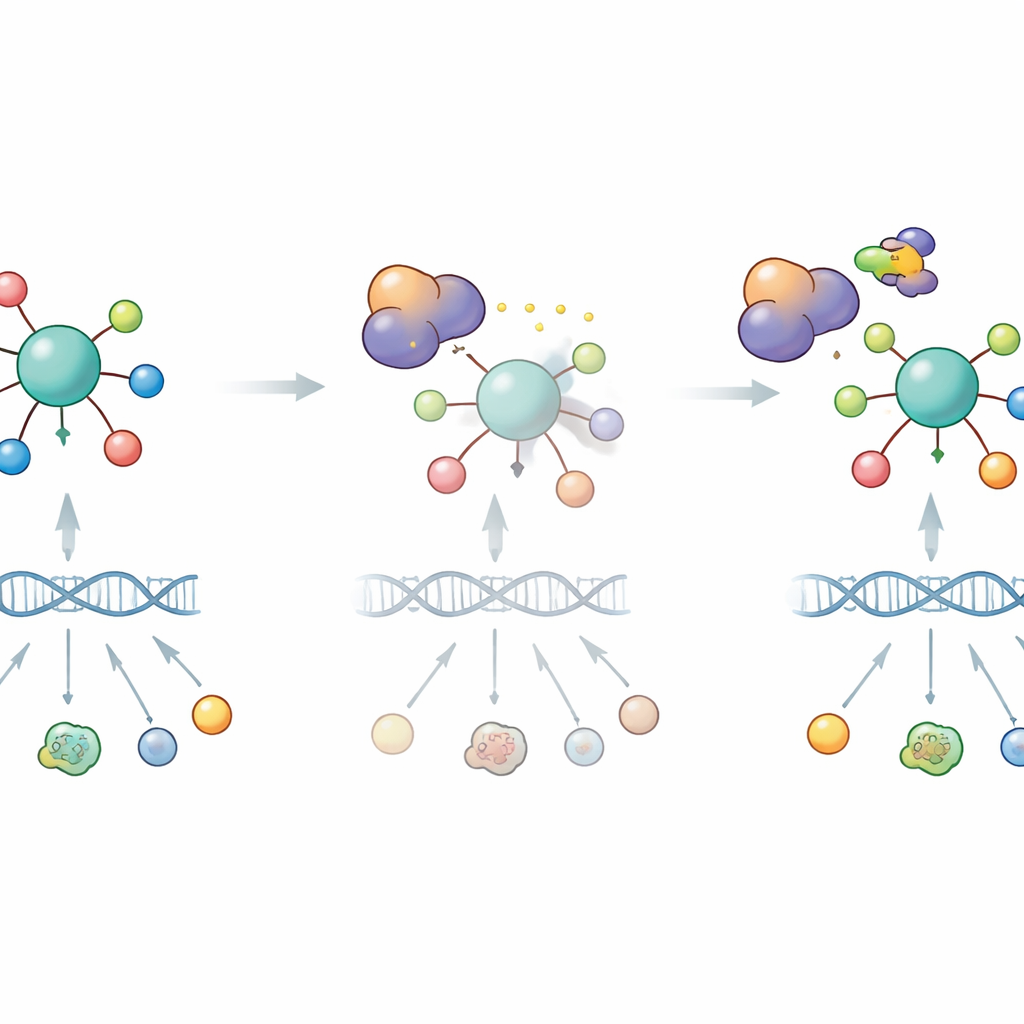
Convertir una debilidad en una oportunidad terapéutica
Puesto que DYRK1B limita la actividad de un supresor tumoral, los autores probaron si bloquear DYRK1B podría restaurar el papel protector de RFX7 y sensibilizar a las células cancerosas frente a la quimioterapia. En células de cáncer de pulmón diseñadas para sobreproducir DYRK1B, dos inhibidores distintos de DYRK1 reactivaron RFX7, aumentaron los niveles de proteínas supresoras tumorales y revirtieron el efecto atenuante de DYRK1B sobre las respuestas mediadas por p53. Un compuesto especializado que degrada selectivamente las quinasas DYRK1 también hizo que las células fueran más vulnerables a la muerte inducida por doxorrubicina, y este efecto de quimiosensibilización se redujo cuando RFX7 estaba ausente. En conjunto, estos hallazgos sugieren que muchos tumores pueden explotar DYRK1B para mitigar la señalización supresora tumoral p53–RFX7, y que dirigir farmacológicamente DYRK1B podría ayudar a reactivar esta vía. Para los pacientes, esto plantea la posibilidad de que futuros inhibidores de DYRK1B, usados junto con la quimioterapia estándar, podrían inclinar la balanza dentro de las células tumorales de vuelta hacia la muerte celular en lugar de la supervivencia.
Cita: Wilms, G., Schwandt, K., Düsterhöft, S. et al. The protein kinase DYRK1B is a p53 target gene and functions as a negative feedback regulator of the transcription factor RFX7. Cell Death Dis 17, 386 (2026). https://doi.org/10.1038/s41419-026-08660-x
Palabras clave: señalización de p53, quinasa DYRK1B, supresor tumoral RFX7, respuesta al estrés en el cáncer, quimiosensibilización