Clear Sky Science · de
Die Proteinkinase DYRK1B ist ein p53-Zielgen und fungiert als negativer Rückkopplungsregulator des Transkriptionsfaktors RFX7
Warum das für die Krebstherapie wichtig ist
Die meisten Krebserkrankungen weisen Schäden an dem berühmten „Wächter des Genoms“, einem Protein namens p53, auf. Dieser Wächter hilft normalerweise Zellen, eine Pause einzulegen, ihre DNA zu reparieren oder sich selbst zu zerstören, wenn etwas schiefgeht. Die hier zusammengefasste Studie enthüllt einen neuen Mechanismus, mit dem Krebszellen dieses Schutzsystem über ein anderes Protein, eine Kinase namens DYRK1B, abschwächen können, und zeigt, dass die Hemmung von DYRK1B Tumorzellen gegenüber Chemotherapie verwundbarer machen könnte.
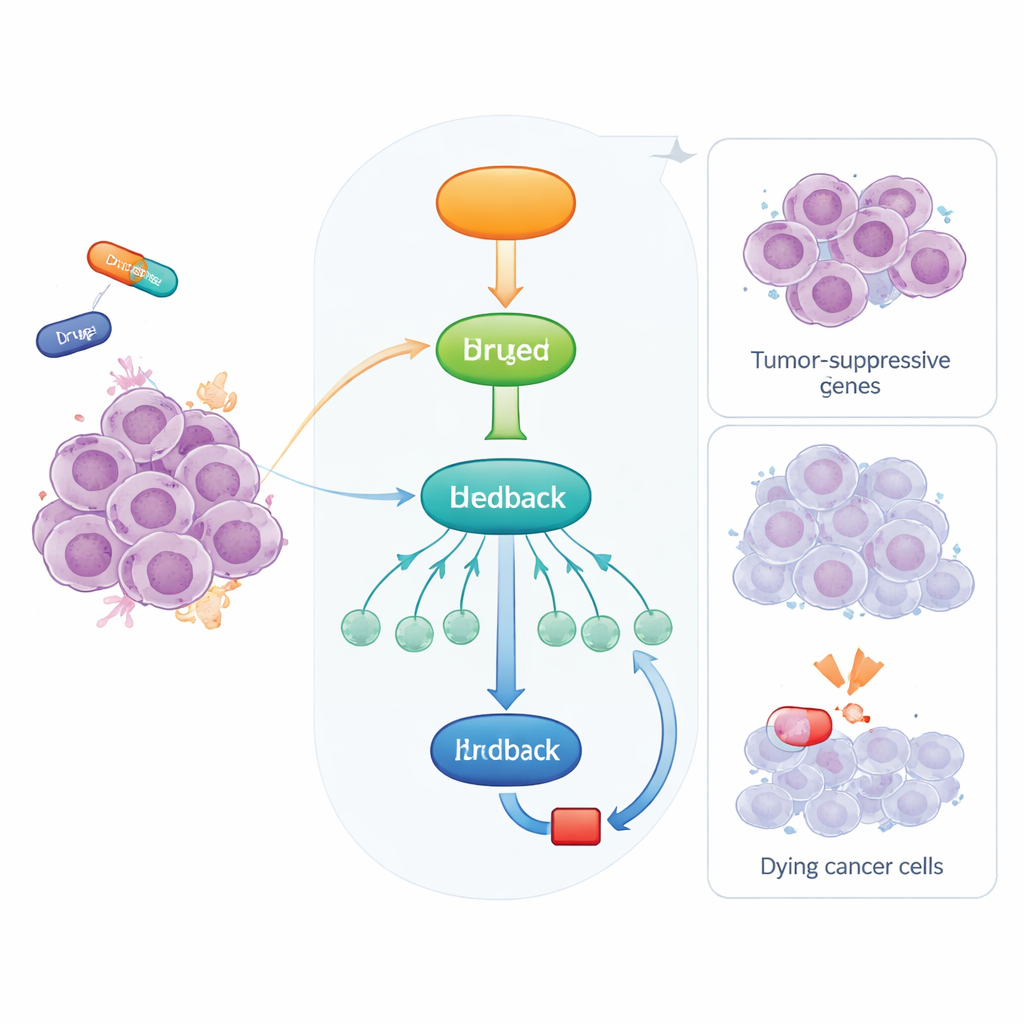
Ein eingebautes Alarmsystem in unseren Zellen
Wenn Zellen Stress ausgesetzt sind, etwa durch DNA-Schäden oder Probleme bei der Herstellung neuer Zellbestandteile, wird p53 aktiviert und startet ein Notprogramm. Das geschieht vorwiegend durch das An- oder Abschalten anderer Gene. Einige dieser Gene stoppen direkt den Zellzyklus oder lösen den Zelltod aus, doch p53 wirkt auch über zusätzliche Steuerproteine, die selbst zahlreiche nachgeschaltete Gene regulieren. Eines davon ist RFX7, ein Transkriptionsfaktor, der sich kürzlich als wichtiger Tumorsuppressor herauskristallisiert hat. RFX7 hilft, ein Genetzwerk zu aktivieren, das das Tumorwachstum hemmt, und ist bei menschlichen Krebserkrankungen häufig gestört oder abgeschwächt.
Eine überlebensfördernde Kinase im Fokus
DYRK1B ist ein Enzym, das anderen Proteinen Phosphatgruppen anhängt und dadurch ihr Verhalten verändert. Frühere Arbeiten zeigten, dass DYRK1B Krebszellen hilft, schwierige Bedingungen zu überstehen, einen niedrigen Aktivitätszustand beizubehalten und DNA-Schäden zu reparieren. Es wird in mehreren soliden Tumoren häufig in ungewöhnlich hohen Mengen gefunden, und seine Blockade kann Krebszellen in experimentellen Modellen empfindlicher gegenüber Chemotherapie oder Strahlung machen. Im Vergleich zu vielen anderen krebsrelevanten Enzymen blieb DYRK1B jedoch lange wenig verstanden und galt als „dunkle Kinase“. Die neue Studie hatte zum Ziel, zu klären, wie DYRK1B gesteuert wird und wie es in die umfassendere stressgesteuerte Schaltkreise passt, die von p53 kontrolliert werden.
Von p53 über RFX7 zu DYRK1B
Die Forscher behandelten verschiedene Krebszelllinien mit zwei Chemotherapeutika, Doxorubicin und Actinomycin D, die beide p53 aktivieren. Sie beobachteten, dass die DYRK1B-Spiegel nach der Behandlung stark anstiegen, während das nahe verwandte DYRK1A dies nicht tat. Mit einem Wirkstoff namens Nutlin-3a, der p53 einschaltet, ohne DNA-Schäden zu verursachen, bestätigten sie, dass DYRK1B immer dann induziert wird, wenn p53 aktiviert ist. Wurde p53 genetisch entfernt, verschwand dieser Anstieg von DYRK1B, und Analysen von Tumordaten von Patientinnen und Patienten zeigten, dass die DYRK1B‑Expression in vielen Krebsarten tendenziell mit den p53‑Spiegeln korreliert. Das Team zeigte dann, dass diese Induktion indirekt erfolgt: p53 stimuliert zuerst RFX7, und RFX7 wiederum erhöht DYRK1B. Das Entfernen von RFX7 oder die Unfähigkeit von RFX7, in den Zellkern zu gelangen, reduzierte die DYRK1B‑Induktion stark, und die DYRK1B‑Spiegel stiegen sowohl auf RNA‑ als auch auf Proteinebene an, was eine echte Genaktivierung bestätigte.
Eine molekulare Bremse auf einen Tumorsuppressor
Einmal produziert, bleibt DYRK1B nicht untätig. Die Studie zeigt, dass DYRK1B in Zellen physisch mit RFX7 assoziiert und dieses modifiziert. Wenn p53 RFX7 aktiviert, geht das Protein in eine Form über, die mit starker Genaktivität einhergeht. Die Hemmung von DYRK1B mit kleinen Molekülen oder dessen Abbau mithilfe eines gezielten Degraders verstärkt diese aktive Form von RFX7, während eine Überexpression von DYRK1B RFX7 wieder in einen weniger aktiven Zustand zurückführt und die Produktion mehrerer von RFX7 gesteuerter Tumorsuppressorproteine, darunter PDCD4, dämpft. Biochemische Experimente zeigen, dass DYRK1B die C‑Terminal‑Region von RFX7 phosphoryliert, wodurch es sich anders in Gelelektrophorese verschiebt und seine transkriptionelle Potenz verliert. Im Kern bildet DYRK1B eine negative Rückkopplungsschleife: p53 schaltet RFX7 ein, RFX7 erhöht DYRK1B, und DYRK1B bremst dann RFX7 wieder.
Eine Schwäche in eine therapeutische Chance verwandeln
Weil DYRK1B die Aktivität eines Tumorsuppressors einschränkt, prüften die Autoren, ob die Blockade von DYRK1B RFX7s Schutzfunktion wiederherstellen und Krebszellen gegenüber Chemotherapie empfindlicher machen kann. In Lungenkrebszellen, die so verändert waren, dass sie DYRK1B überproduzieren, konnten zwei verschiedene DYRK1‑Inhibitoren RFX7 reaktivieren, die Spiegel tumorsuppressiver Proteine erhöhen und den dämpfenden Effekt von DYRK1B auf p53‑gesteuerte Antworten umkehren. Eine spezialisierte Verbindung, die DYRK1‑Kinasen selektiv abbaut, machte Zellen zudem anfälliger für doxorubicin‑induzierten Zelltod, und dieser chemosensibilisierende Effekt war reduziert, wenn RFX7 fehlte. Insgesamt legen diese Befunde nahe, dass viele Tumoren DYRK1B nutzen könnten, um die p53–RFX7‑Tumorsuppressor‑Signalgebung abzuschwächen, und dass die pharmakologische Zielrichtung auf DYRK1B dazu beitragen könnte, diesen Weg wieder zu reaktivieren. Für Patientinnen und Patienten eröffnet dies die Möglichkeit, dass künftig eingesetzte DYRK1B‑Inhibitoren in Kombination mit Standardchemotherapie das Gleichgewicht in Krebszellen wieder zugunsten des Zelltods statt des Überlebens verschieben könnten.
Zitation: Wilms, G., Schwandt, K., Düsterhöft, S. et al. The protein kinase DYRK1B is a p53 target gene and functions as a negative feedback regulator of the transcription factor RFX7. Cell Death Dis 17, 386 (2026). https://doi.org/10.1038/s41419-026-08660-x
Schlüsselwörter: p53-Signalübertragung, DYRK1B-Kinase, RFX7-Tumorsuppressor, Stressantwort bei Krebs, Chemo-Sensibilisierung