Clear Sky Science · en
Single-cell and bulk transcriptomic analyses uncover immune subtypes associated with programmed cell death features in intrahepatic cholangiocarcinoma
Why This Liver Cancer Study Matters
Intrahepatic cholangiocarcinoma is a fast-growing cancer that arises from the small bile ducts inside the liver and is often discovered too late for surgery. Doctors know that some patients live much longer or respond better to new immunotherapies than others, but it has been difficult to predict who will benefit. This study uses advanced genetic profiling and single-cell analysis to show how different forms of "programmed" cell death and the immune system interact in these tumors, and builds a practical score that could help tailor treatments and guide clinical decisions.
Looking Inside Liver Tumors Cell by Cell
The researchers gathered large datasets of gene activity from hundreds of tumor samples, including both bulk tissue and single cells, from public resources and a large clinical cohort. They focused on 2,701 genes linked to 21 types of programmed cell death – the built-in ways cells can self-destruct when damaged or abnormal. By comparing tumor tissue with nearby non-cancerous liver, they narrowed this list to 87 genes that were both misregulated in cancer and potentially important for patient survival. These genes were involved in known cancer pathways and forms of cell death such as apoptosis and necroptosis, and also showed characteristic patterns of mutations and chromosome changes in liver bile duct tumors. 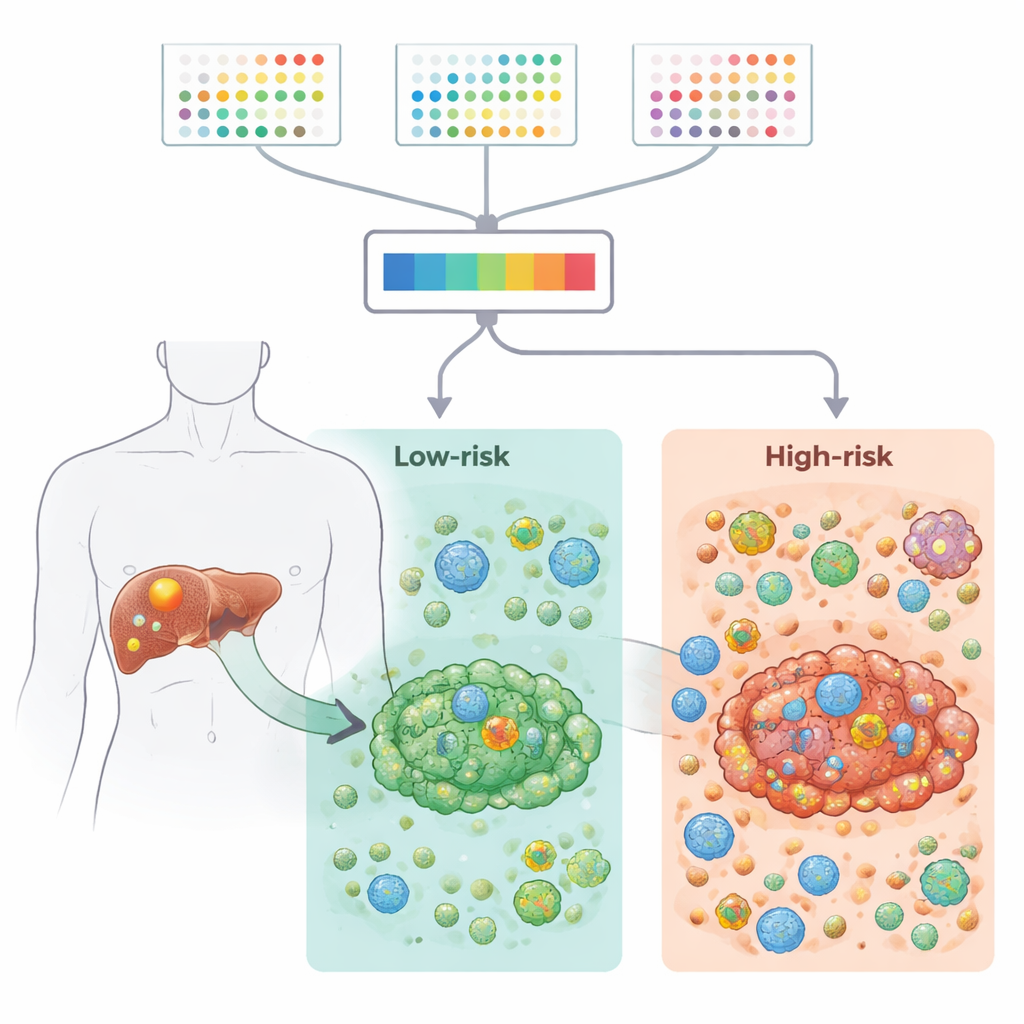
Building a Risk Score from Nine Key Genes
To convert this complex information into something clinically useful, the team tested 117 combinations of machine-learning methods and survival models. The best-performing approach, which combined stepwise Cox regression with random survival forests, distilled the information down to nine genes. Each patient received a risk score based on how strongly these genes were turned on or off in their tumor. Across several independent groups of patients, this nine-gene signature reliably separated people into high- and low-risk groups with clearly different survival times. High-risk tumors were enriched for stress and inflammation-related pathways, while low-risk tumors showed more active metabolic and detoxification pathways, suggesting fundamental biological differences between the two groups.
Immune Neighborhoods Inside the Tumor
The study then zoomed in on the tumor microenvironment – the mix of immune cells, support cells, and connective tissue that surround and infiltrate the cancer. Using multiple computational tools, the authors showed that high-risk tumors had higher immune and stromal scores, stronger inflammatory signals, and more immunosuppressive features, including increased regulatory T cells that can blunt anti-tumor responses. Single-cell RNA sequencing confirmed that these regulatory T cells and fibroblasts were more abundant in high-risk tumors and engaged in dense networks of cell–cell communication. In contrast, low-risk tumors had stronger collagen-related signaling and a more balanced, less suppressive environment. Together, these findings suggest that the nine-gene pattern is tightly linked to how "friendly" or "hostile" the local immune neighborhood is toward the cancer. 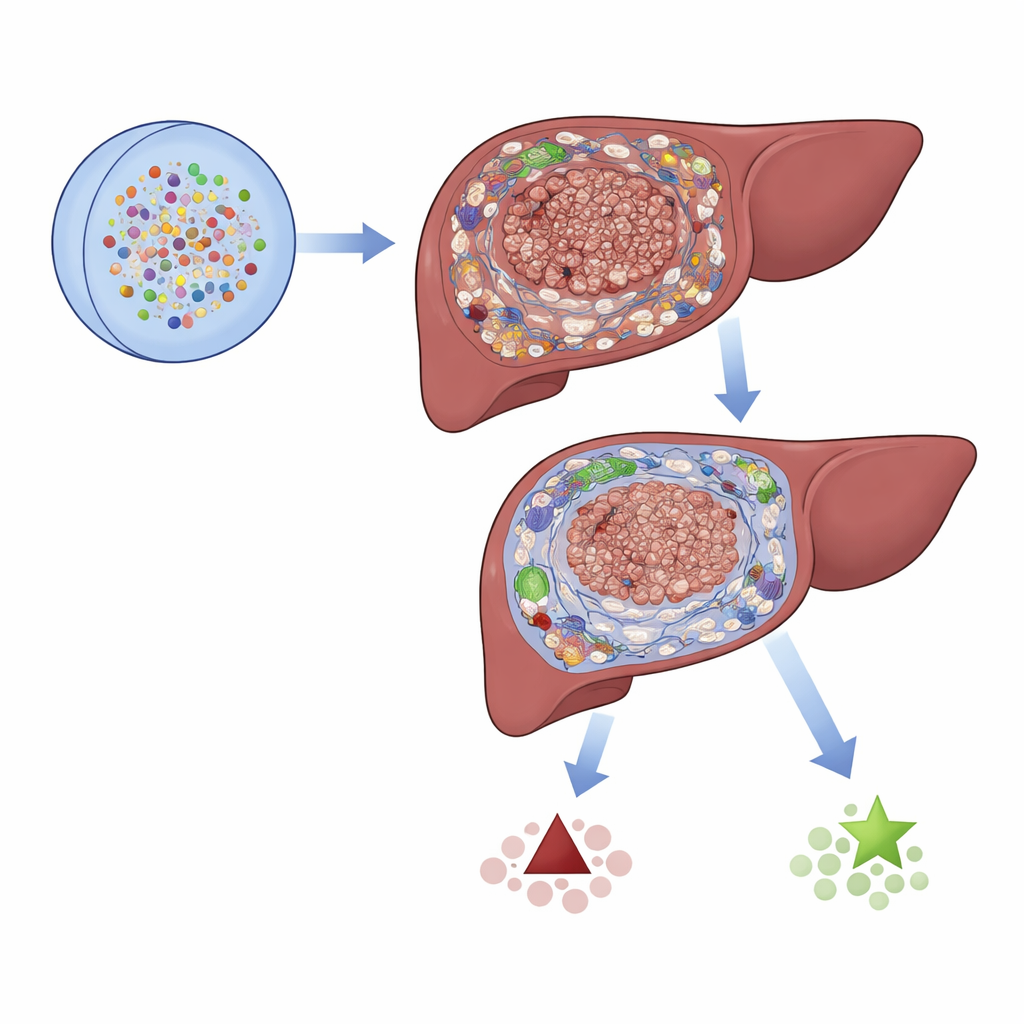
Guiding Immunotherapy and Drug Choices
Recognizing that immunotherapy is becoming central to cancer care, the researchers created a separate "PCD score" from the same nine genes to predict response to immune checkpoint blockade. In three independent patient cohorts treated with such drugs (from other cancer types with available data), higher PCD scores were consistently associated with better responses, implying that the score captures features of a tumor environment that make immunotherapy more likely to work. The team also used drug sensitivity databases and computer docking simulations to predict which medicines might be more effective in different risk groups. High-risk patients appeared more sensitive to certain targeted agents, including those that interfere with TGF-beta and mTOR signaling, offering possible combination strategies to overcome immune suppression.
From Complex Biology to Practical Tools
To help translate these findings to the clinic, the authors built a nomogram – a visual risk calculator – that combines the gene-based risk score with routine clinical information like stage and vascular invasion. This tool accurately predicted one-, three-, and four-year survival in their main patient cohort. Although the work still needs prospective testing in larger, treatment-defined groups of intrahepatic cholangiocarcinoma patients, it provides a blueprint for how patterns of cell death–related genes can be used to classify tumors, understand their immune ecosystems, and guide therapy. For patients, the long-term promise is more precise forecasts and better-matched treatments based on the unique genetic and immune fingerprint of their tumor.
Citation: Zhang, T., Dou, D., Liu, Q. et al. Single-cell and bulk transcriptomic analyses uncover immune subtypes associated with programmed cell death features in intrahepatic cholangiocarcinoma. Sci Rep 16, 13678 (2026). https://doi.org/10.1038/s41598-026-44332-8
Keywords: intrahepatic cholangiocarcinoma, programmed cell death, tumor immune microenvironment, single-cell RNA sequencing, immunotherapy response