Clear Sky Science · de
Einzelzell- und Bulk-Transkriptomanalysen decken Immunsubtypen auf, die mit Merkmalen programmierter Zellsterblichkeit bei intrahepatischem Gallengangskarzinom assoziiert sind
Warum diese Leberkrebsstudie wichtig ist
Das intrahepatische Cholangiokarzinom ist ein schnell wachsender Tumor, der aus den kleinen Gallengängen in der Leber entsteht und häufig zu spät für eine Operation entdeckt wird. Ärztinnen und Ärzte wissen, dass einige Patientinnen und Patienten deutlich länger leben oder besser auf neue Immuntherapien ansprechen als andere, aber es war schwer vorherzusagen, wer profitieren wird. Diese Studie nutzt fortgeschrittene genetische Profile und Einzelzellanalyse, um zu zeigen, wie verschiedene Formen des „programmierten“ Zelltods und das Immunsystem in diesen Tumoren zusammenwirken, und entwickelt eine praktische Punktzahl, die helfen könnte, Behandlungen anzupassen und klinische Entscheidungen zu unterstützen.
Blick in Lebertumoren, Zelle für Zelle
Die Forschenden sammelten umfangreiche Datensätze zur Genaktivität aus Hunderten Tumorproben, sowohl aus Bulk-Gewebe als auch aus Einzelzellen, aus öffentlichen Quellen und einer großen klinischen Kohorte. Sie konzentrierten sich auf 2.701 Gene, die mit 21 Formen des programmierten Zelltods verbunden sind – den eingebauten Mechanismen, mit denen Zellen sich selbst zerstören können, wenn sie beschädigt oder abnormal sind. Durch den Vergleich von Tumorgewebe mit benachbarter nicht-krebsartiger Leber schränkten sie diese Liste auf 87 Gene ein, die sowohl fehlreguliert im Krebs als auch potenziell wichtig für das Überleben der Patientinnen und Patienten waren. Diese Gene waren in bekannten Krebswegen und Zelltodformen wie Apoptose und Nekroptose beteiligt und zeigten charakteristische Muster von Mutationen und Chromosomenveränderungen in Tumoren der lebereigenen Gallengänge. 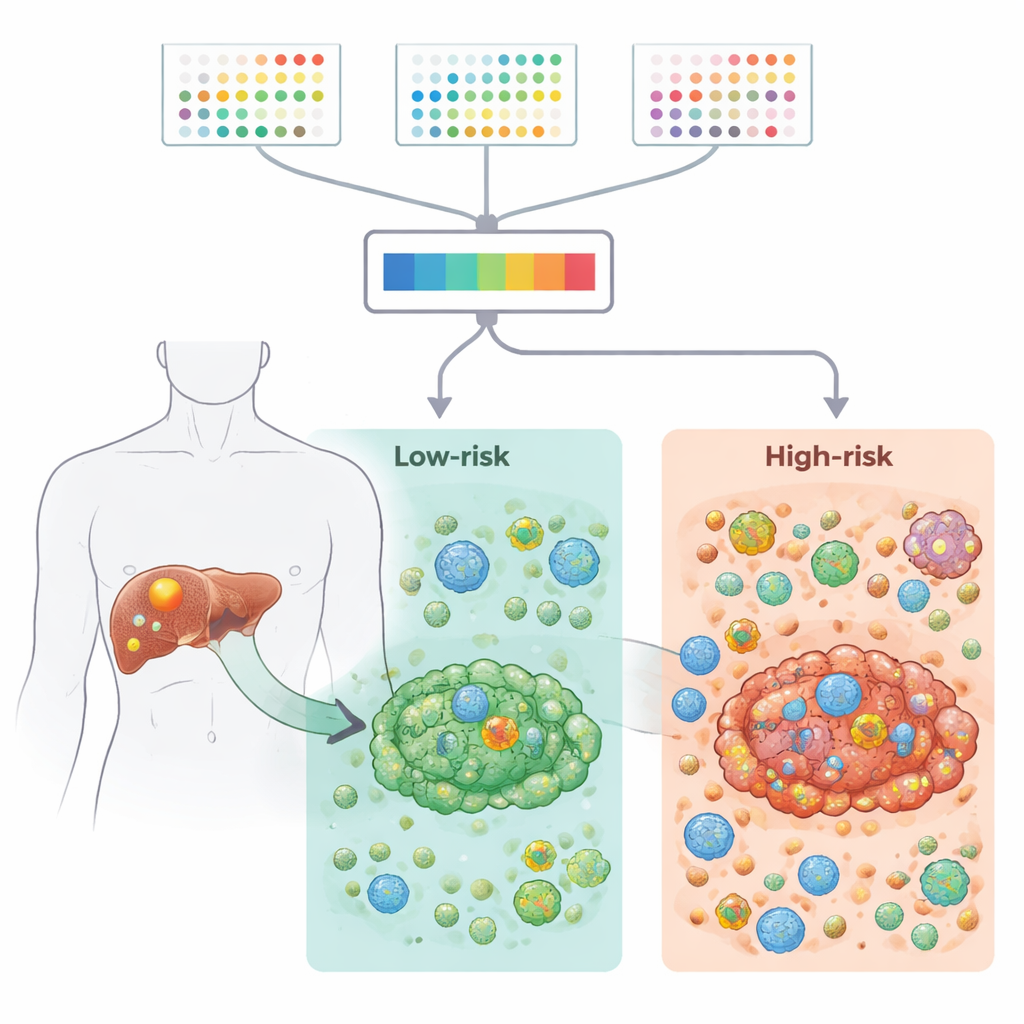
Entwicklung eines Risikoscores aus neun Schlüsselgenen
Um diese komplexen Informationen in etwas klinisch Nützliches zu überführen, testete das Team 117 Kombinationen aus Machine-Learning-Verfahren und Überlebensmodellen. Der leistungsstärkste Ansatz, der schrittweise Cox-Regression mit Random Survival Forests kombinierte, destillierte die Informationen auf neun Gene. Jede Patientin und jeder Patient erhielt einen Risikoscore basierend darauf, wie stark diese Gene im Tumor ein- oder ausgeschaltet waren. In mehreren unabhängigen Patientengruppen trennte diese Neun-Gen-Signatur zuverlässig in Hoch- und Niedrigrisikogruppen mit deutlich unterschiedlichen Überlebenszeiten. Hochrisikotumoren waren angereichert für Stress- und Entzündungswege, während Niedrigrisikotumoren aktivere Stoffwechsel- und Entgiftungswege zeigten, was auf grundlegende biologische Unterschiede zwischen den Gruppen hindeutet.
Immune Nachbarschaften innerhalb des Tumors
Die Studie zoome dann in das tumorassoziierte Mikroumfeld – die Mischung aus Immunzellen, Stütz- und Bindegewebe, die den Krebs umgibt und infiltriert. Mit mehreren rechnerischen Werkzeugen zeigten die Autorinnen und Autoren, dass Hochrisikotumoren höhere Immun- und Stromascores, stärkere Entzündungssignale und mehr immunsuppressive Merkmale aufwiesen, darunter vermehrte regulatorische T-Zellen, die anti-tumorale Reaktionen dämpfen können. Einzelzell-RNA-Sequenzierung bestätigte, dass diese regulatorischen T-Zellen und Fibroblasten in Hochrisikotumoren häufiger vorkamen und dichte Kommunikationsnetzwerke zwischen Zellen bildeten. Im Gegensatz dazu wiesen Niedrigrisikotumoren stärkere Kollagen-assoziierte Signale und ein ausgeglicheneres, weniger unterdrückendes Umfeld auf. Zusammen deuten diese Befunde darauf hin, dass das Neun-Gen-Muster eng mit der Frage verknüpft ist, wie „freundlich“ oder „feindlich“ die lokale Immunumgebung dem Tumor gegenübersteht. 
Leitlinien für Immuntherapie und Arzneimittelauswahl
In dem Bewusstsein, dass die Immuntherapie zunehmend zentral in der Krebsbehandlung wird, erstellten die Forschenden einen separaten „PCD-Score“ aus denselben neun Genen, um das Ansprechen auf eine Blockade von Immun-Checkpoints vorherzusagen. In drei unabhängigen Patientenkohorten, die mit solchen Medikamenten behandelt wurden (aus anderen Krebsarten mit verfügbaren Daten), gingen höhere PCD-Scores konsequent mit besseren Antworten einher, was darauf hindeutet, dass der Score Merkmale der Tumorumgebung erfasst, die Immuntherapien wahrscheinlich erfolgreicher machen. Das Team nutzte außerdem Arzneimittelsensitivitätsdatenbanken und computergestützte Docking-Simulationen, um vorherzusagen, welche Wirkstoffe in den verschiedenen Risikogruppen effektiver sein könnten. Hochrisikopatientinnen und -patienten schienen auf bestimmte zielgerichtete Wirkstoffe empfindlicher zu sein, darunter solche, die in TGF‑beta- und mTOR-Signalisierung eingreifen, und bieten mögliche Kombinationsstrategien, um Immunsuppression zu überwinden.
Von komplexer Biologie zu praktischen Instrumenten
Um die Überführung dieser Ergebnisse in die Klinik zu erleichtern, entwickelten die Autorinnen und Autoren ein Nomogramm – einen visuellen Risikorechner –, das den genbasierten Risikoscore mit routinemäßigen klinischen Informationen wie Tumorstadium und Gefäßinvasion kombiniert. Dieses Werkzeug sagte das Ein-, Drei- und Vier-Jahres-Überleben in ihrer Hauptkohorte zuverlässig voraus. Zwar bedarf die Arbeit noch prospektiver Tests in größeren, behandlungsdefinierten Kohorten von Patientinnen und Patienten mit intrahepatischem Cholangiokarzinom, doch liefert sie einen Bauplan dafür, wie Muster zelltodbezogener Gene zur Klassifikation von Tumoren, zum Verständnis ihrer Immunökosysteme und zur Therapiewahl genutzt werden können. Für Patientinnen und Patienten ist die langfristige Perspektive präzisere Prognosen und besser angepasste Therapien auf Basis des einzigartigen genetischen und immunologischen Fingerabdrucks ihres Tumors.
Zitation: Zhang, T., Dou, D., Liu, Q. et al. Single-cell and bulk transcriptomic analyses uncover immune subtypes associated with programmed cell death features in intrahepatic cholangiocarcinoma. Sci Rep 16, 13678 (2026). https://doi.org/10.1038/s41598-026-44332-8
Schlüsselwörter: intrahepatisches Gallengangskarzinom, programmierter Zelltod, tumorimmunes Mikroenvironment, Einzelzell-RNA-Sequenzierung, Immuntherapieansprechen