Clear Sky Science · en
SynRXN: An Open Benchmark and Curated Dataset for Computational Reaction Modeling
Why Smarter Chemistry Data Matter
Modern chemistry increasingly relies on computers to help design new medicines, materials, and everyday chemicals. But even the smartest algorithms are only as good as the data they learn from. Today, reaction data are scattered across patents, papers, and lab notebooks in messy, inconsistent formats, making it hard to judge whether competing computer tools are truly improving. This article introduces SynRXN, an open, carefully curated collection of reaction datasets and tests designed to give researchers a common playing field for evaluating how well their methods work in planning and understanding chemical syntheses.
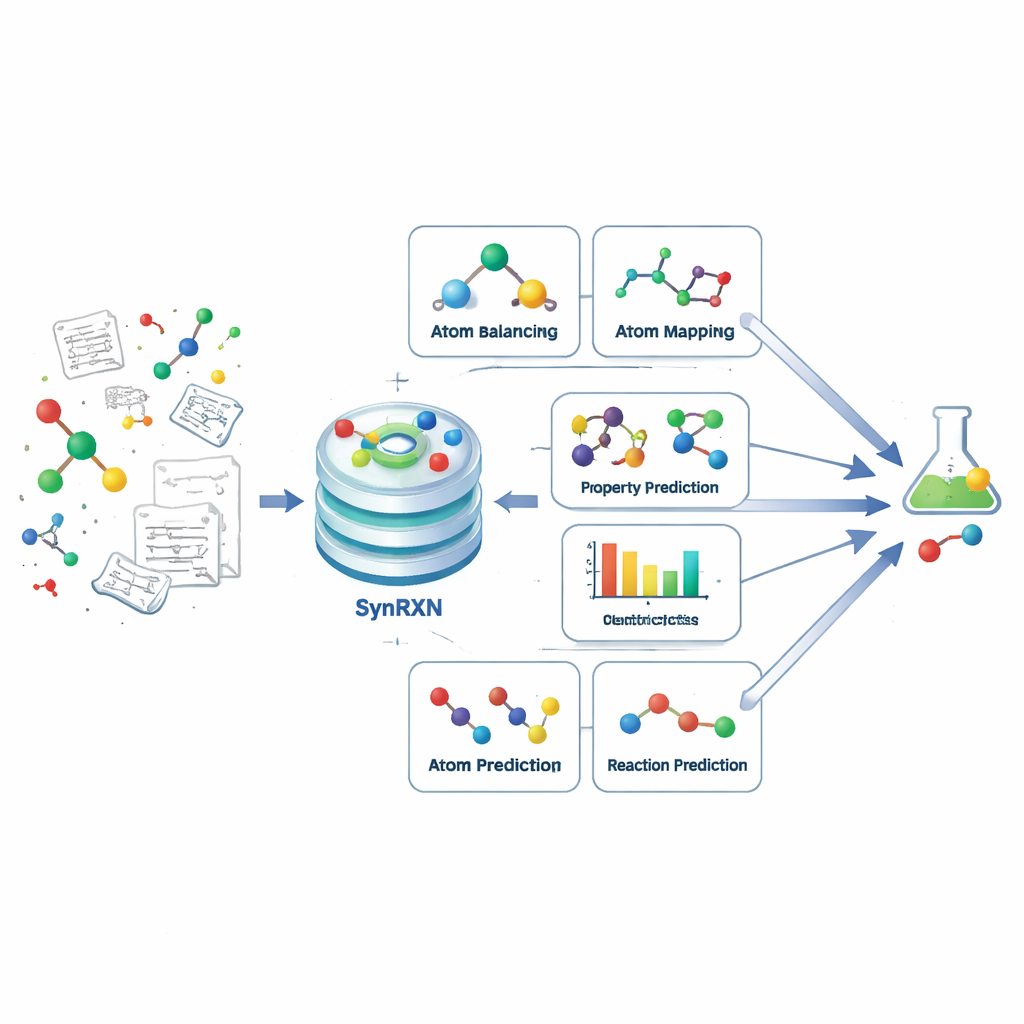
Breaking a Complex Problem into Clear Steps
Computer-aided synthesis planning aims to help chemists figure out how to make a target molecule from simpler building blocks. Instead of treating this as one monolithic challenge, SynRXN breaks it into five concrete task families that mirror what chemists do in practice. First comes fixing and completing reaction records (rebalancing), then tracking how individual atoms move from starting materials to products (atom-to-atom mapping), then assigning each reaction to a category (reaction classification), then predicting useful numerical properties such as reaction yields or energy barriers (reaction property prediction), and finally forecasting which products or starting materials are likely for a given transformation (synthesis prediction). By defining precise inputs, outputs, and evaluation measures for each of these steps, SynRXN turns a tangled workflow into a set of well-posed problems.
Cleaning Up Noisy Chemical Records
Real-world reaction data are often incomplete: patents and electronic lab notebooks may leave out solvents, counterions, or side products, or list impossible atom counts. SynRXN tackles this in its reaction rebalancing task by starting from widely used patent-derived datasets and deliberately constructing examples with specific types of errors, such as missing non-carbon atoms or components missing on one or both sides of the equation. It then uses a hybrid rule- and graph-based method to restore balance, keeping only corrections with very high confidence and manually verifying the final test set. For atom-to-atom mapping, SynRXN gathers both organic and biochemical reactions from several trusted sources and subjects them to strict, automated checks to ensure that molecules are valid and consistently represented before comparing how well different mapping tools track the fate of each atom.
Organizing Reactions into Classes and Numbers
Beyond simply knowing whether a reaction is balanced, chemists care about what type of transformation it is and how well it performs. SynRXN therefore assembles several tiers of reaction classification tasks, ranging from broad categories down to fine-grained differences in mechanism, and includes both organic reactions from patents and enzyme-catalyzed reactions from biology. It pairs these labels with standardized ways to split data into training, validation, and test sets, and with agreed-upon performance metrics. For numerical targets, SynRXN collects datasets of reaction yields, energy barriers, rates, and other properties from the literature and public repositories. All are run through a consistent cleaning pipeline, and simple reference models are provided so users can quickly tell whether a new method is genuinely better than a reasonable baseline.
Making Reaction Predictions Fair and Reproducible
For the synthesis prediction task, SynRXN focuses on single-step reactions where a given set of reactants leads to one or more products, or where the goal is to work backward from a product to plausible starting materials. It bundles three influential patent-derived corpora that many groups already use, but re-exports them with deterministic, transparent splits and common evaluation scripts. Under the hood, all SynRXN datasets follow the same tabular structure with stable reaction identifiers, standardized molecule encodings, and explicit license tags. A machine-readable manifest records file locations, checksums, column names, and counts, enabling anyone to regenerate the same curated tables on a different machine or at a later date using scripted build recipes.
What This Means for Future Chemical Discovery
In practical terms, SynRXN does not introduce a new predictive model; instead, it provides the scaffolding needed to compare existing and future models fairly. By harmonizing reaction data from many sources, enforcing strict quality checks, and publishing open, versioned benchmarks with reference results, SynRXN allows researchers to pinpoint which parts of the synthesis-planning pipeline are working well and where they fail. For chemists and data scientists alike, this means that claims of improved performance can be grounded in shared tests rather than bespoke, opaque datasets, accelerating trustworthy progress toward computer tools that truly assist real-world chemical design.
Citation: Phan, TL., Nguyen Song, NN. & Stadler, P.F. SynRXN: An Open Benchmark and Curated Dataset for Computational Reaction Modeling. Sci Data 13, 625 (2026). https://doi.org/10.1038/s41597-026-07260-w
Keywords: computer-aided synthesis planning, reaction benchmarking, chemical reaction datasets, machine learning for chemistry, reaction prediction