Clear Sky Science · en
Telomere shortening in laminopathic dilated cardiomyopathy
Why heart cell aging matters
Many people with inherited heart muscle diseases develop serious problems such as irregular heartbeats, heart failure, and the need for transplants at relatively young ages. This study asks a simple but important question: do the heart cells in these patients age faster at the most basic level of their DNA? By looking closely at tiny structures called telomeres—protective caps on chromosomes—the researchers explore how a fault in a key scaffolding protein of the cell’s nucleus might speed up cellular wear and tear in the heart.
Protective caps at the ends of our DNA
Every chromosome in our cells ends in a stretch of repeated DNA that acts like the plastic tip of a shoelace, preventing fraying. These telomeres normally shorten slowly as we age, and very short telomeres are considered a hallmark of cellular aging. In the heart, most muscle cells stop dividing soon after birth, so their telomere length is expected to remain fairly steady. Earlier work by this group showed that certain genetic heart diseases break this rule, with telomeres becoming unexpectedly short. Here, the focus is on a family of rare disorders called laminopathies, caused by mutations in the LMNA gene, which builds the internal shell of the cell nucleus and helps maintain its shape and stiffness.
Heart tissue that looks older than it should
The team first examined heart biopsy samples from patients with different forms of laminopathy who had undergone heart transplantation, and compared them with hearts from people without cardiac disease. Using a fluorescent technique that measures telomere signals in individual heart muscle cell nuclei, they found that laminopathy hearts had on average 38 percent lower telomere levels than controls. This pattern held across several clinical subtypes, and patients with one form of the disease, called LGMD1B, showed especially rapid telomere loss with age. In contrast, telomere levels in healthy hearts were remarkably stable over time, reinforcing the idea that LMNA mutations drive an abnormal acceleration of telomere shortening in heart cells.
Modeling diseased heart cells in a dish
To probe cause and effect, the researchers created induced pluripotent stem cells from a patient carrying a frameshift LMNA mutation, then used gene-editing tools to generate matched cell lines with two normal copies, one mutated copy, or no functional copies of LMNA. In the stem-cell state, adding back a normal LMNA copy increased telomere length, while complete loss of LMNA produced only modest telomere gains, highlighting a delicate balance between nuclear structure and telomere maintenance. When these stem cells were turned into beating heart cells in the lab, the picture changed: lines with little or no normal LMNA showed pronounced telomere loss, supporting the idea that the mechanical stress of repeated contraction makes nuclear integrity especially important for preserving telomeres in heart muscle.
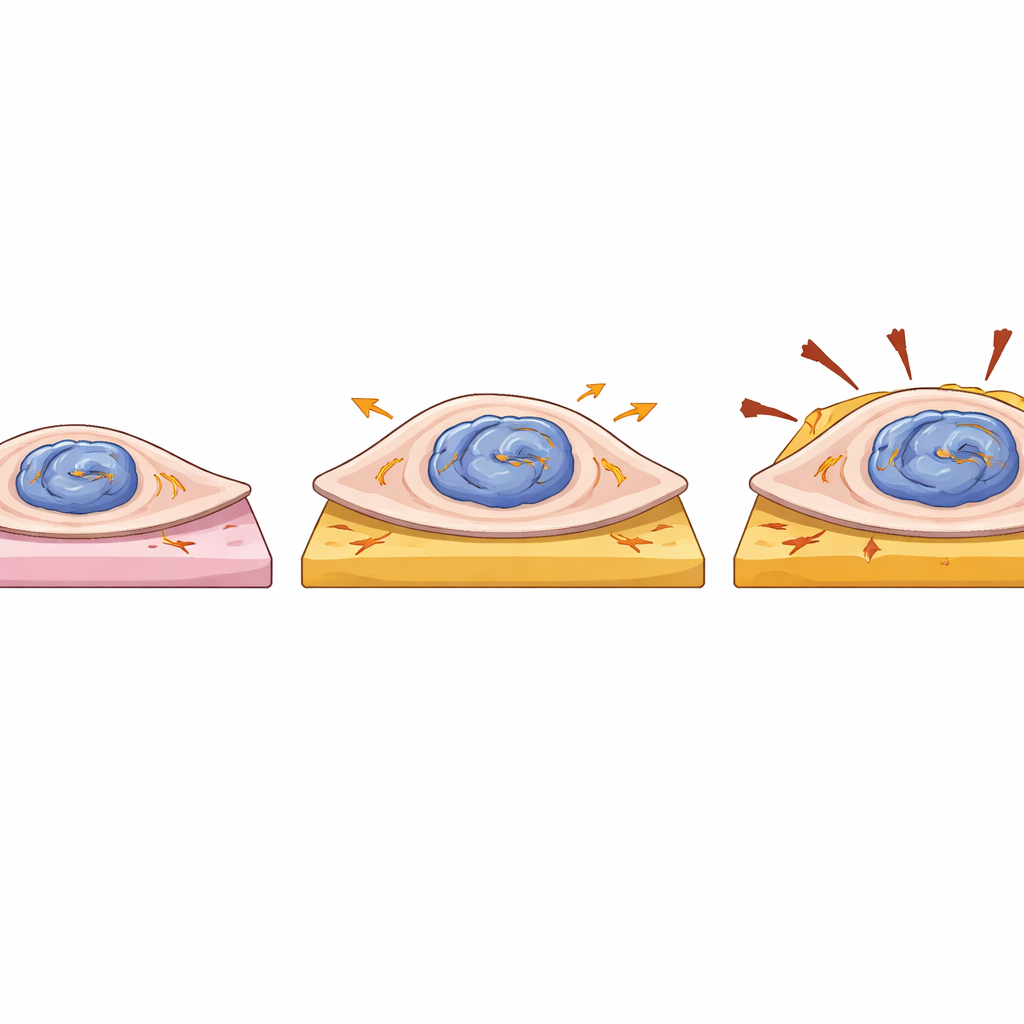
Stronger yet more fragile heart cells
The team also asked how the mutation affects the physical behavior of single heart cells. They placed individual patient-derived and corrected cells onto tiny, soft or stiff gel pads that mimic healthy and scarred heart tissue, and measured how hard each cell pulled as it contracted. Cells carrying one mutant LMNA copy were larger in area and generated greater overall force and strain energy than their corrected counterparts, particularly on softer gels. However, this increased force did not come with faster contraction, and these cells showed subtle irregularities in beat rate, hinting at disturbed internal signaling. Because the average movement of the gel surface was similar between groups, the extra force seems to come mainly from increased cell size, not more efficient machinery. Together with nucleus deformation, this suggests that mutant LMNA makes heart cells bulkier and more mechanically sensitive, while at the same time eroding their telomere protection.
Evidence from a humanized mouse model
To see whether these findings hold in a whole organism, the researchers turned to mice engineered to carry a human LMNA mutation that produces progerin, the altered protein responsible for a premature-aging syndrome. These mice developed early signs of dilated cardiomyopathy, with reduced pumping performance. Their heart muscle cells also had significantly shorter telomeres and showed diminished shortening during each beat, pointing to impaired contractile function. These animal results mirror the human tissue and cell culture data, strengthening the link between LMNA mutation, telomere loss, and failing heart muscle.
What this means for future care
In plain terms, this work shows that certain inherited defects in the scaffolding of the cell nucleus can make heart muscle cells both work harder and age faster, as seen by the loss of their chromosome end-caps. The degree of telomere shortening varies with the specific LMNA mutation and cell type, which may help explain why patients with similar gene changes can have different disease severity. By establishing that telomere erosion is a consistent feature of laminopathy-related heart disease, the study points to telomere protection and nuclear support as potential new targets for therapies aimed at slowing or preventing heart failure in these patients.
Citation: Chang, A.C.Y., Pardon, G., Chang, A.C.H. et al. Telomere shortening in laminopathic dilated cardiomyopathy. npj Regen Med 11, 16 (2026). https://doi.org/10.1038/s41536-026-00462-1
Keywords: laminopathy, telomeres, dilated cardiomyopathy, heart muscle cells, cellular aging