Clear Sky Science · de
Die Beiträge von TLR2, TLR8 und TLR3 zur direkten und antikörperabhängigen Verstärkung der Infektion mit Dengue‑Virus Serotyp 2
Warum diese Forschung wichtig ist
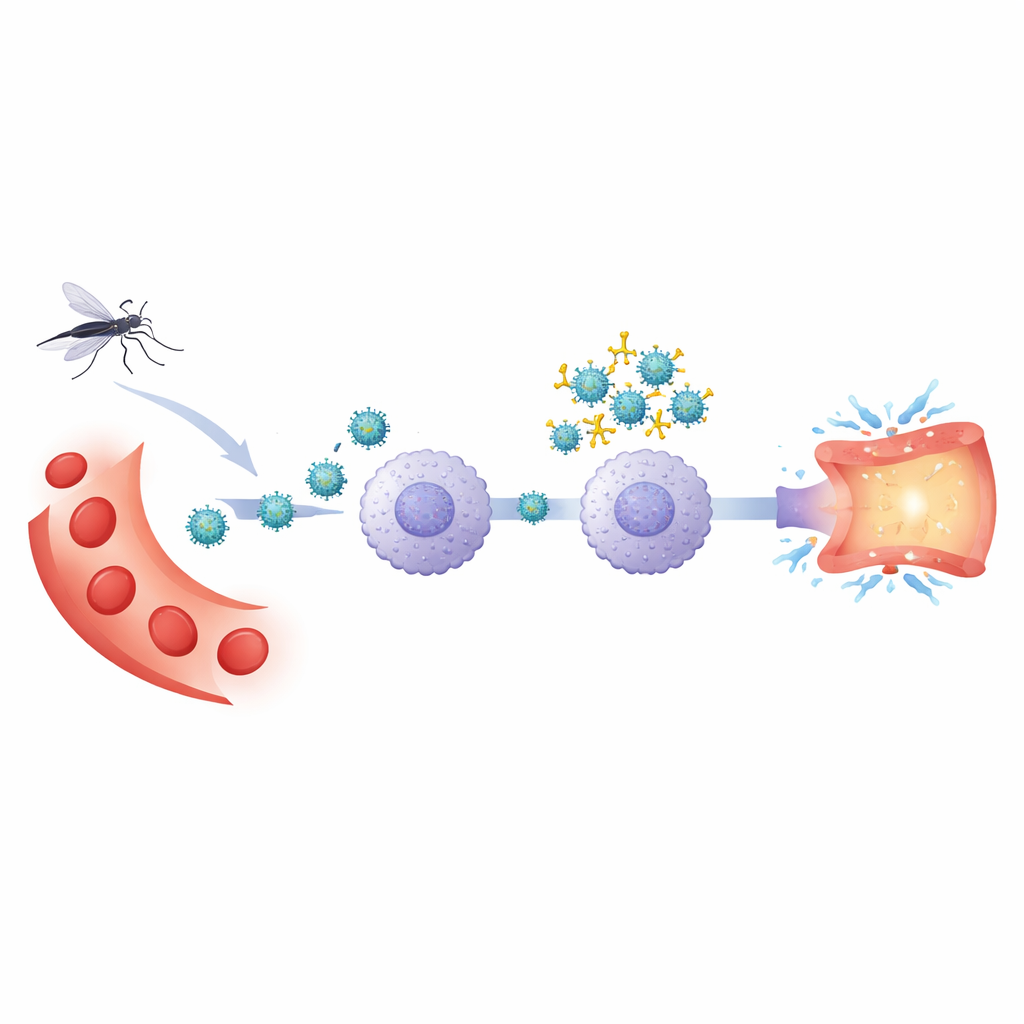
Denguefieber ist eine durch Mücken übertragene Krankheit, die von einer starken, grippeähnlichen Erkrankung bis zu lebensbedrohlichen Blutungen und Schock reichen kann. Personen, die sich ein zweites Mal mit Dengue infizieren, haben ein erhöhtes Risiko für schwere Verläufe — besonders dann, wenn vorhandene Antikörper statt Schutz die Infektion fördern und dem Virus helfen, Immunzellen zu infizieren. Diese Studie identifiziert zentrale Moleküle auf Immunzellen, die dieses gefährliche "Aufschaukeln" von Infektion und Entzündung ermöglichen, und weist auf neue Ansätze hin, um die schwersten Formen von Dengue zu verhindern.
Wenn nützliche Antikörper schädlich werden
Nach einer ersten Dengue‑Infektion behält der Körper Antikörper, die das Virus erkennen. Bei einer späteren Infektion mit einem anderen Dengue‑Typ neutralisieren diese Antikörper das Virus möglicherweise nicht vollständig. Stattdessen können sie das Virus umhüllen und es über Antikörperrezeptoren in bestimmte weiße Blutkörperchen leiten — ein Vorgang, der als antikörperabhängige Verstärkung (antibody‑dependent enhancement) bezeichnet wird. Die Autoren verwendeten ein experimentelles System auf Basis menschlicher Blutzellen, um diese verstärkte Infektion mit einer normalen Infektion durch Dengue‑Virus Typ 2 zu vergleichen. Im Fokus standen Monozyten, eine Art Immunzelle, die sowohl Wirtszelle für das Virus ist als auch entzündungsfördernde Substanzen freisetzt, die Blutgefäße schädigen können.
Wichtige Torwächter auf Immunzellen
Das Team untersuchte mehrere Rezeptoren — molekulare "Andockstationen" — auf Monozyten. Einer war CD32, ein Rezeptor, der Antikörper erkennt, die an das Virus gebunden sind. Weitere waren Sensoren, die typischerweise Erreger detektieren, darunter TLR2 an der Zelloberfläche sowie TLR3 und TLR8 in intrazellulären Kompartimenten, ebenso wie ein Signalenzym namens SYK, das Rezeptoren mit nachgeschalteten Reaktionen verbindet. Durch gezieltes Blockieren dieser Moleküle mit Antikörpern oder kleinen Inhibitoren prüften sie, welche tatsächlich für das Eindringen des Virus und den darauf folgenden Entzündungsstoß erforderlich sind.
TLR2 als wesentliche Eintrittshilfe
Die Experimente zeigten, dass beim antibody‑vermittelten Eindringen des Dengue‑Virus das Blockieren von CD32 oder SYK die Infektionsrate auf das Niveau ohne Antikörper zurückbrachte. Auffällig war, dass die Blockade von TLR2 noch weiter ging: Sie verhinderte die Infektion nahezu vollständig, sowohl mit als auch ohne Antikörper. Das weist darauf hin, dass TLR2 nicht nur als Alarmgeber fungiert, sondern auch eine entscheidende Helferrolle beim Eindringen des Virus in Monozyten unter allen Bedingungen spielt. Ein weiteres Molekül, CD14, unterstützte diesen Prozess, während andere vorgeschlagene Co‑Rezeptoren wie TLR1, TLR6, CD36 oder LILRB1 in diesem Modell keine erkennbare Rolle spielten.
Vom Viruseintritt zur Gefäßschädigung
Schweres Dengue zeichnet sich durch undichte Blutgefäße aus. Um Ereignisse in Monozyten mit Effekten auf Blutgefäße zu verbinden, sammelten die Forschenden das Medium aus infizierten Blutkultur‑Proben und gaben es an im Labor gezüchtete menschliche Endothelzellen von Venen weiter. Diese Gefäßzellen reagierten, indem sie Oberflächenmarker hochfuhren, die Entzündung und Leckage fördern — jedoch nur, wenn sie den von infizierten Monozyten freigesetzten Substanzen ausgesetzt wurden, nicht durch Virus‑Antikörper‑Gemische allein. Die antibody‑verstärkte Infektion veranlasste Monozyten, noch mehr dieser aktivierenden Signale zu produzieren als die direkte Infektion. Das Blockieren von CD32, SYK oder TLR2 in den Blutzellen reduzierte diese Endothelaktivierung stark und senkte zudem die Mengen an antiviralen und entzündlichen Molekülen wie Interferonen und TNF‑alpha.
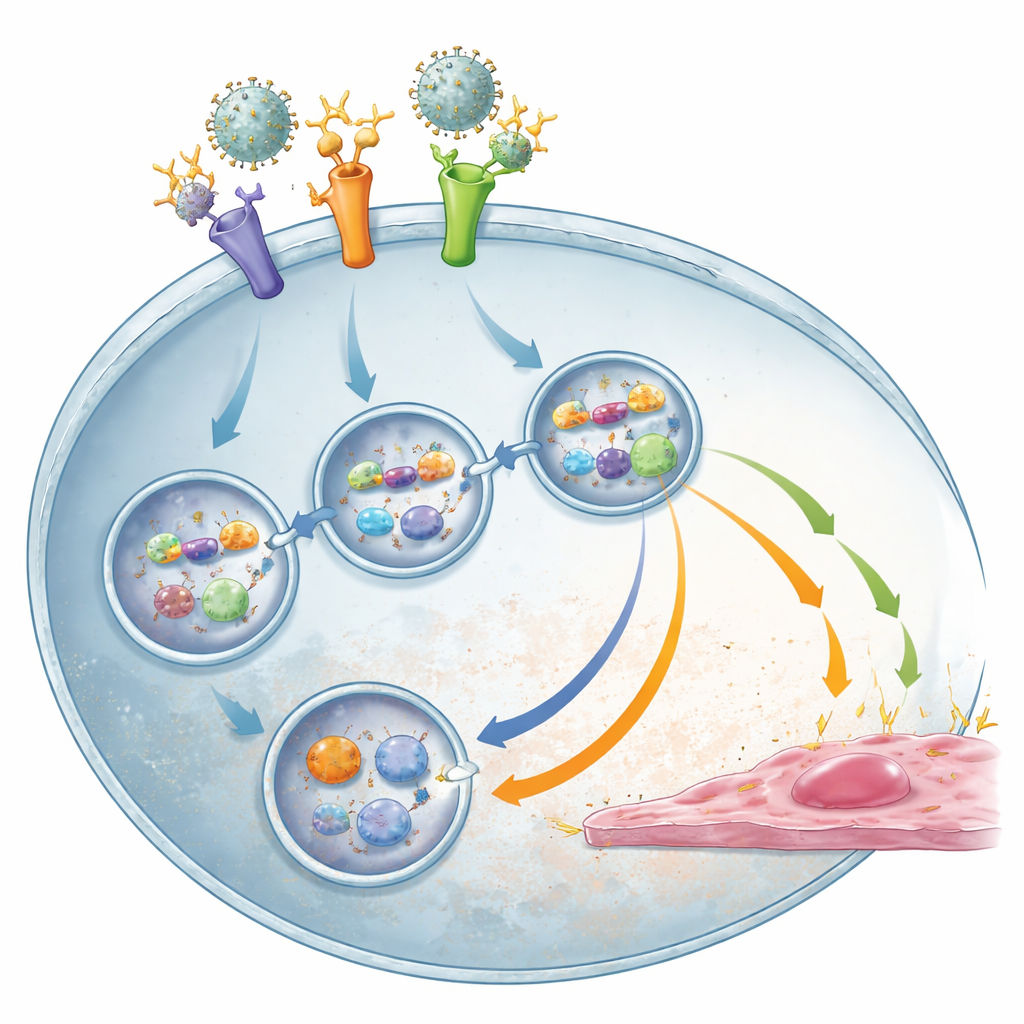
Versteckte virale RNA entfacht den Sturm
Um herauszufinden, was den Entzündungssturm tatsächlich auslöst, prüften die Forschenden, ob nicht infektiöse Viruspartikel Monozyten noch aktivieren können. Wurden die viralen Nukleinsäuren durch UV‑Behandlung zerstört, induzierten die Partikel keine gefäßaktivierenden Signale mehr, obwohl TLR2 sie weiterhin binden konnte. Das deutete auf virale RNA als entscheidenden Auslöser hin. Tatsächlich führten das Blockieren der intrazellulären RNA‑Sensoren TLR3 und insbesondere TLR8 dazu, dass Monozyten deutlich weniger entzündliche und antivirale Substanzen produzierten — vor allem unter antibody‑verstärkten Bedingungen — ohne die Infektionsrate der Zellen zu verringern. Das spricht für einen zweistufigen Prozess: Zuerst hilft TLR2, Virus‑Antikörper‑Komplexe in die Zelle zu bringen; sobald virale RNA freigelegt ist, erkennen TLR3 und TLR8 diese und treiben die schädliche Immunantwort an, wobei SYK als gemeinsamer Signalhub wirkt.
Was das für die Bekämpfung schweren Dengues bedeutet
Insgesamt schlägt die Studie eine klare Abfolge von Ereignissen vor, die eine sekundäre Dengue‑Infektion mit schwerer Erkrankung verknüpft. Antikörper aus einer früheren Infektion können das Dengue‑Virus über CD32 in Monozyten einschleusen, doch ein erfolgreicher Infektionsverlauf in diesen Zellen hängt auch von TLR2 ab. Im Inneren der Zelle setzen TLR3 und TLR8 zusammen mit SYK ein kräftiges entzündliches und antivirales Programm in Gang, das Gefäße aktiviert und schädigt. Indem TLR2, TLR3, TLR8 und SYK als zentrale Akteure sowohl beim Viruseintritt als auch bei der schädlichen Entzündung identifiziert werden, liefert diese Arbeit potenzielle Wirkstoffziele, die eines Tages helfen könnten, einen milden Dengueverlauf daran zu hindern, sich in eine lebensbedrohliche Form zu verwandeln.
Zitation: ter Ellen, B.M., Punekar, M., Castillo, J.A. et al. The contributions of TLR2, TLR8 and TLR3 to direct and antibody-dependent enhancement of dengue virus serotype 2 infection. npj Viruses 4, 24 (2026). https://doi.org/10.1038/s44298-026-00190-9
Schlüsselwörter: Dengue‑Virus, antikörperabhängige Verstärkung, angeborene Immunität, Toll‑like‑Rezeptoren, Gefäßleckage