Clear Sky Science · de
Ontologiegestützte Assoziationsregel-Analyse für Beziehungen zwischen biomedizinischen Entitäten: Integration hierarchischen Wissens zur Verbesserung der Gen‑Krankheits‑Entdeckung
Warum verdeckte Gen–Krankheits‑Verbindungen wichtig sind
Die moderne Medizin ist zunehmend darauf angewiesen, herauszufinden, welche Gene mit welchen Krankheiten verbunden sind. Solche Verknüpfungen können aufzeigen, warum Krankheiten entstehen, neue Wirkstoffziele vorschlagen und Personen mit erhöhtem Risiko identifizieren. Die meisten Computerwerkzeuge suchen jedoch nur nach Genen und Krankheiten, die im selben Satz oder Artikel gemeinsam erwähnt werden, und übersehen viele subtile, aber wichtige Zusammenhänge. Diese Studie stellt eine neue Methode vor, um die biomedizinische Literatur zu durchforsten, die auf expertengefertigte Wissenshierarchien zurückgreift und darauf abzielt, sowohl bekannte als auch übersehene Gen–Krankheits‑Beziehungen verlässlicher aufzudecken.
Vom Rohtext zu Kandidatenverknüpfungen
Die Autoren beginnen damit, eine große Sammlung wissenschaftlicher Artikel aus PubMed zu erfassen und jeden Artikel in Sätze zu zerlegen. Jeder Satz wird wie ein kleiner „Korb“ von Elementen behandelt, der einen oder mehrere Gen‑Namen und einen oder mehrere Krankheitsnamen enthalten kann. Mithilfe etablierter Data‑Mining‑Algorithmen (Apriori, FP‑Growth und Eclat) durchforsten sie Millionen solcher Körbe, um Gen–Krankheits‑Paare zu finden, die häufiger gemeinsam auftreten, als es durch Zufall zu erwarten wäre. Dieser erste Schritt, genannt entity‑specific association, erfasst die direkten Ko‑Vorkommen, auf die sich die meisten vorhandenen Werkzeuge stützen. Er zeigt bereits Tausende potenzieller Verbindungen, tendiert jedoch weiterhin zu gut untersuchten Genen und häufigen Krankheiten, die die Literatur dominieren.
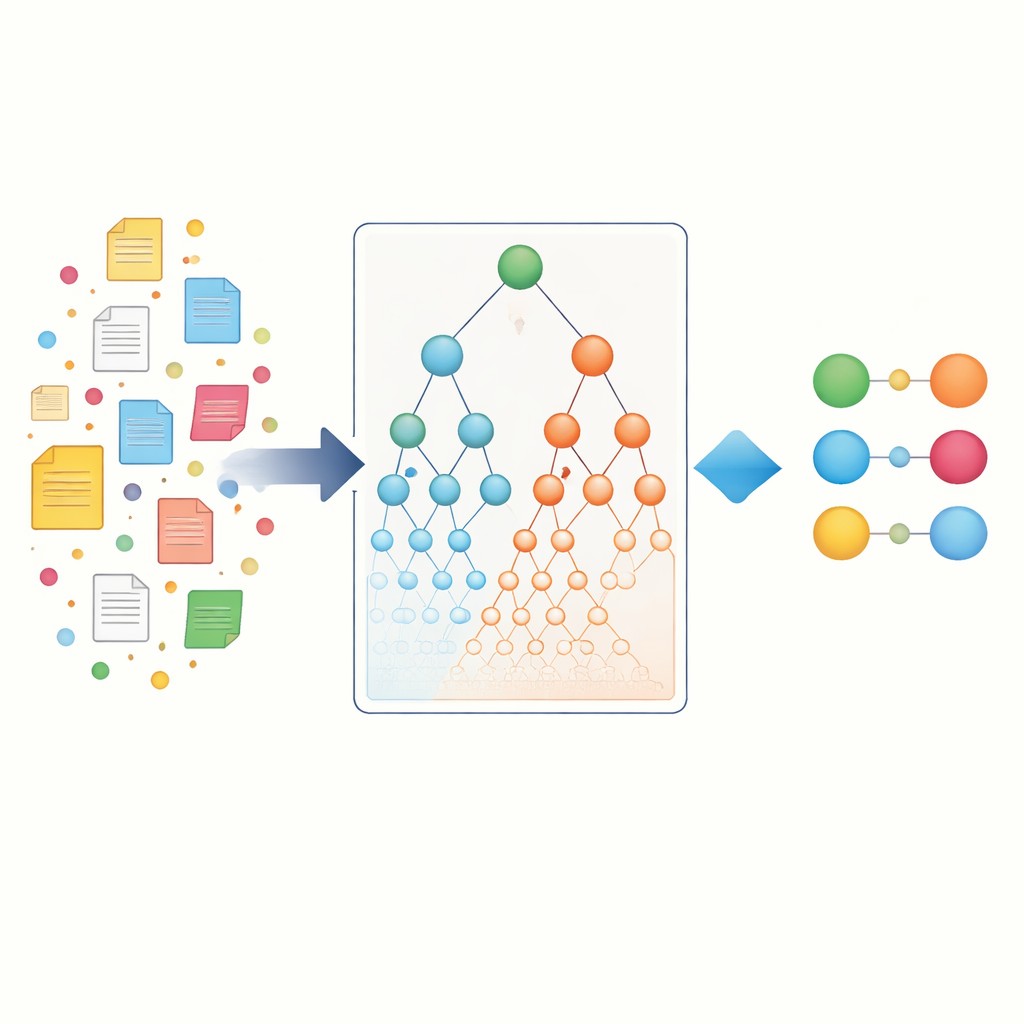
Biologische Hierarchien als Landkarte nutzen
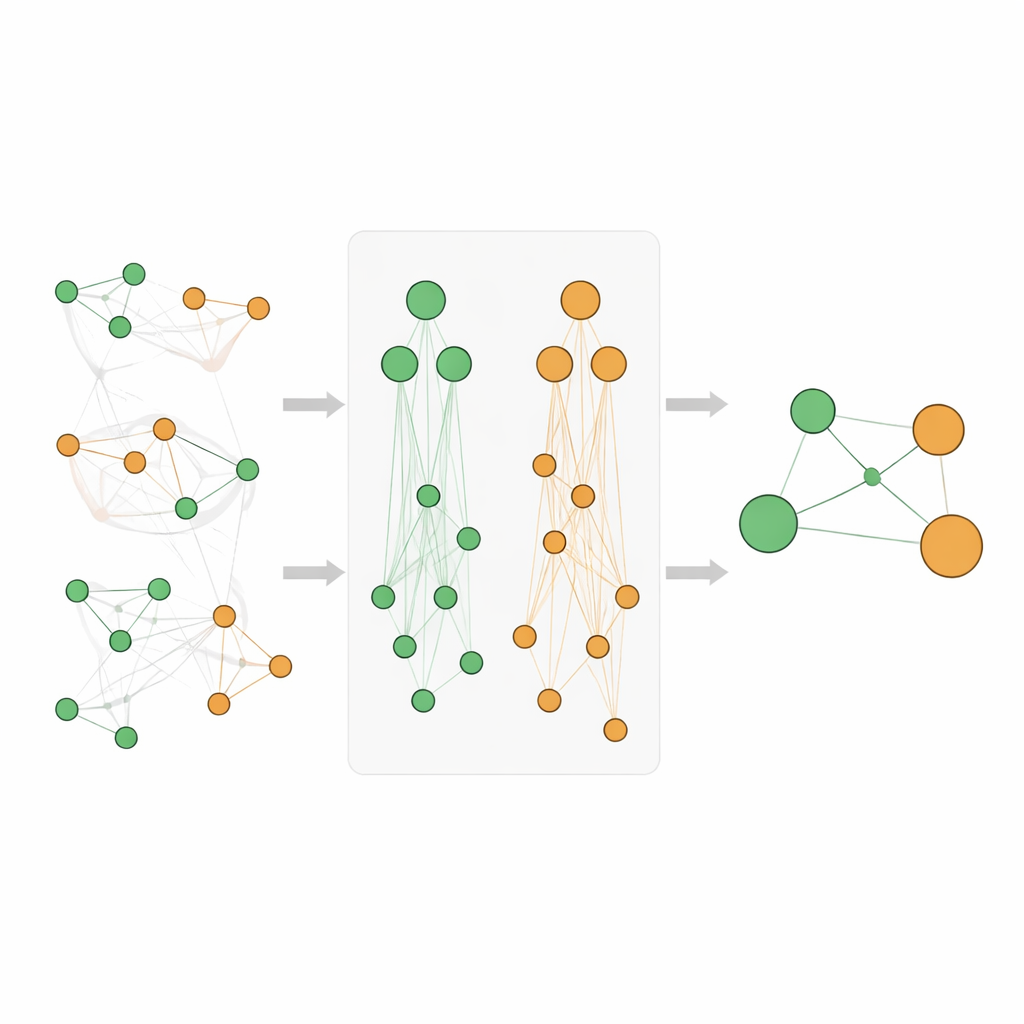
Um über einfaches Wortzählen hinauszugehen, wenden sich die Forschenden biologischen „Landkarten“ zu, den Ontologien. Die Gene Ontology beschreibt, was Gene tun und wo sie in der Zelle wirken, während die Disease Ontology Krankheiten in Familien und Subtypen organisiert. In diesen Hierarchien stehen spezifische Begriffe, etwa eine seltene Epilepsie, unter allgemeineren Elternbegriffen wie „neurologische Erkrankung“. Die zentrale Idee ist: Wenn ein bestimmtes Gen stark mit einer sehr spezifischen Krankheit verbunden ist und diese Krankheit zu einer größeren Familie gehört, dann hat das Gen vermutlich auch eine Beziehung zu dieser gesamten Familie. Die Autoren formalisieren dies durch die Erstellung hierarchischer Ontologie‑Assoziationen, die Evidenz über Elternebenen sowohl auf Gen‑ als auch auf Krankheitsseite weitergeben und zudem indirekt „Geschwister“-Beziehungen erfassen, die denselben Elternknoten teilen.
Direkte Evidenz mit vererbten Signalen mischen
Einfaches Aufsummieren von Zählungen über mehrere Ebenen der Hierarchie kann Scores verzerren, insbesondere weil sehr allgemeine Begriffe wie „Krebs“ extrem häufig vorkommen. Das Team entwickelt daher ein sorgfältiges Bewertungssystem. Sie verwenden ein Standardmaß aus dem Data Mining, genannt Lift, um zu messen, wie stark ein Gen und eine Krankheit über den Zufall hinaus verknüpft sind, und transformieren diese Werte, um Verzerrungen zu reduzieren und Vergleichbarkeit herzustellen. Ihr neuer Athar Semantic‑Enriched Association (ASEA)‑Score kombiniert drei Komponenten: die direkte Gen–Krankheits‑Verbindung, Verbindungen zwischen dem Gen und breiteren Krankheitsfamilien sowie Verbindungen zwischen breiteren Genfunktionen und Krankheitsfamilien. Zusätzlich wenden sie eine rangbasierte Normalisierung an, sodass Scores über verschiedene Ontologie‑Tiefen hinweg vergleichbar sind und faire Rangfolgen ermöglichen.
Evaluation der Methode gegenüber vertrauenswürdigen Datenbanken
Um zu beurteilen, ob ASEA biologisch sinnvolle Ergebnisse liefert, vergleichen die Autoren ihre am höchsten bewerteten Assoziationen mit Einträgen in expertenkurierten Ressourcen wie der Comparative Toxicogenomics Database und DisGeNET. Sie finden heraus, dass ASEA mehr hochrangige bekannte Assoziationen wiederfindet als die klassischen Algorithmen allein, während gleichzeitig eine reichhaltige Menge zusätzlicher Kandidaten generiert wird. Insgesamt identifiziert ASEA 185 bemerkenswerte Gen–Krankheits‑Paare. Diese werden in vier Kategorien gruppiert: gut etablierte Verbindungen, die bereits in großen Datenbanken vorhanden sind; Verbindungen, die stark durch aktuelle Studien gestützt, aber noch nicht kuratiert sind; Verknüpfungen mit nur schwacher oder verstreuter Datenbankunterstützung; und rein spekulative Assoziationen ohne derzeitige Unterstützung, die als Hypothesen für zukünftige Labor‑ oder klinische Untersuchungen vorgeschlagen werden.
Was das für die zukünftige Medizin bedeutet
Für Nicht‑Fachleute ist die entscheidende Botschaft, dass dieses Rahmenwerk eine intelligentere Methode bietet, die biomedizinische Literatur großflächig zu lesen. Statt nur offensichtliche Nennungen eines Gens und einer Krankheit nebeneinander zu zählen, nutzt es Expertenwissen darüber, wie Gene und Krankheiten in Familien organisiert sind, um vielversprechende, aber seltene Signale zu stärken. Der resultierende ASEA‑Score beweist nicht, dass ein Gen eine Krankheit verursacht, liefert jedoch eine transparente, statistisch fundierte Shortlist von Kandidaten, die Forschende und Kliniker weiter untersuchen können. Langfristig könnte ein ontologie‑bewusstes Mining die Entdeckung von Biomarkern beschleunigen, die Präzisionsmedizin informieren und helfen, die wachsende Flut biomedizinischer Texte in verwertbare medizinische Erkenntnisse zu verwandeln.
Zitation: Naqash, M.A., Amin, M., Uddin, J. et al. Ontology-driven association rule mining for biomedical entity relationships: integrating hierarchical knowledge to improve gene-disease discovery. Sci Rep 16, 13072 (2026). https://doi.org/10.1038/s41598-026-42584-y
Schlüsselwörter: Gen–Krankheits‑Assoziationen, biomedizinisches Text‑Mining, Ontologien, Präzisionsmedizin, Computationale Biologie