Clear Sky Science · zh
通过物理信息几何不变学习实现精确的蛋白质-蛋白质相互作用建模
为何蛋白质配对重要
在每个细胞内部,蛋白质很少单独行动。它们相互碰撞、像拼图块一样结合,形成控制信号、运输货物和抵抗感染的短暂配对。确切弄清两种蛋白在三维中如何相遇,可以揭示疾病的成因以及如何设计新药或抗体。但在实验室中获得这些分子“快照”既缓慢又昂贵,而现有的人工智能工具在数据稀缺时仍容易出错。本研究提出了ProTact,一种旨在弥补这些不足的新计算方法。
当前结构预测工具的局限
像AlphaFold这样的系统已改变了我们预测单个蛋白和某些复合体形状的能力。它们在有许多相关蛋白序列可用时表现最佳,因为模型可以从进化模式中学习。然而,许多重要靶点(包括病毒蛋白、工程蛋白和抗体)缺乏丰富的进化历史。在这些情况下,现有工具有时会误判蛋白如何结合,尤其是在两者原子接触的精确接触点处。作者认为,要克服这一点,模型必须超越序列模式,更加关注蛋白表面的物理形状与契合度。
读取蛋白表面的新方法
ProTact 将每个蛋白视为在空间中相连的残基网络,并侧重于它们外表面如何互补。它使用一个几何编码器,精心设计以确保在旋转或平移蛋白时内部表示保持不变。第二个模块应用受三角学启发的规则来推理由残基组形成的三角形,帮助模型捕捉多体效应,而不仅仅是成对接触。两部分协同产生一张地图,突出显示两种蛋白在复合体形成时哪些残基可能接触。ProTact 可与高质量的实验结构配合使用,也可用于其他工具预测的构象,并且在有序列信息时可以利用它,但不依赖于它。
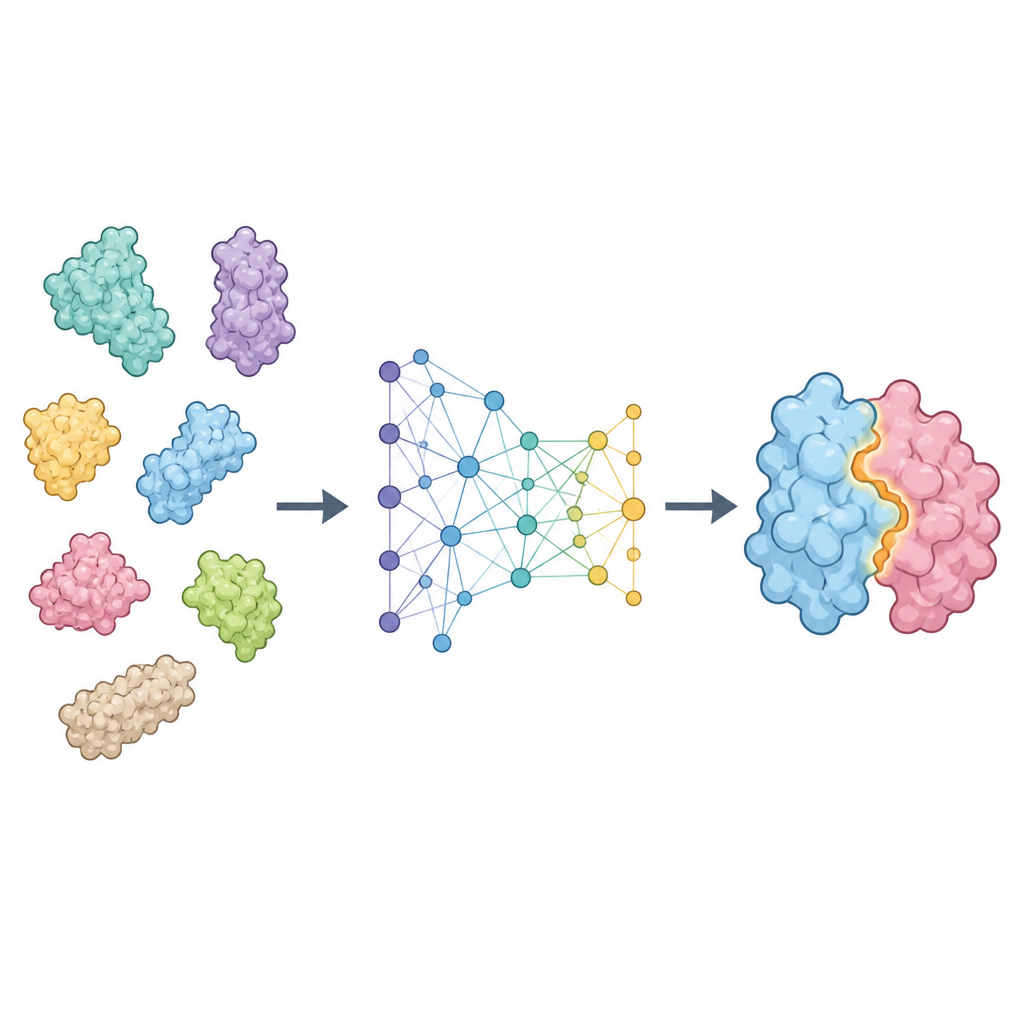
该方法的性能如何
研究人员在若干标准蛋白对集合上测试了ProTact,包括来自社区性评估实验的高难度案例。在这些基准上,ProTact 在识别接触残基方面始终比领先的基于序列和基于结构的方法更准确。例如,在两个广泛使用的数据集中,与现有最佳方法相比,它将常用的精准度分数提高了约三分之一。即使输入结构来自AlphaFold而非实验,因此包含噪声,ProTact 仍然比竞争工具更准确。该方法还处理了大量近期解决的蛋白对的盲测,在对称和非对称复合体上均表现出强劲性能。
从接触图到完整复合体
预测哪些残基接触只是问题的一部分;科学家还希望得到配对的完整三维排列。ProTact 的接触图可以转换为对接构象,即两种蛋白如何结合的完整模型。使用标准的对齐算法,作者表明ProTact 提出的构象比其他基于接触的方法更接近实验确定的复合体。将这些接触预测作为约束加入现有对接程序,可以提高它们在大多数测试对上的准确性。ProTact 还可以对 AlphaFold3 生成的候选复合体重新排序,使其更接近实验质量评分,并改善若干在序列数据稀缺情况下的抗体–抗原实例的结果。
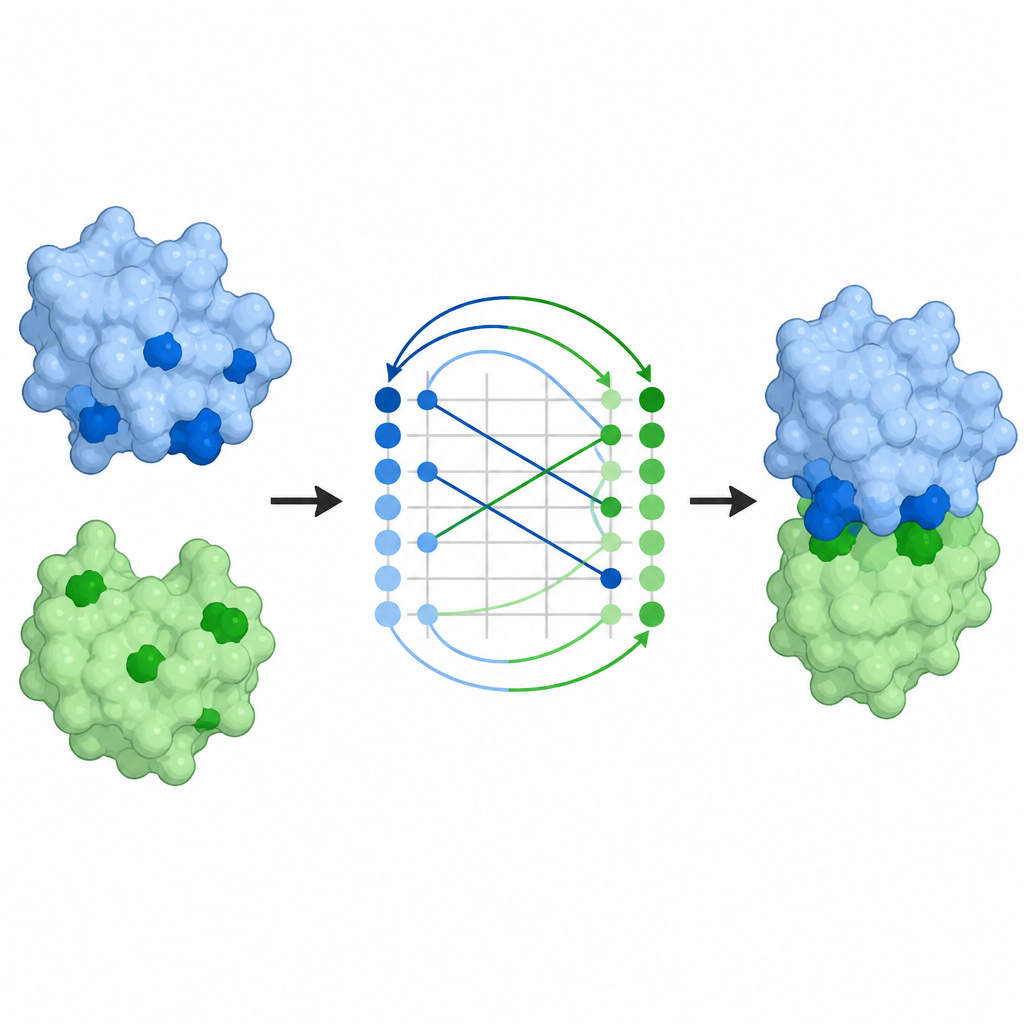
对抗体和药物设计的意义
抗体是特别艰难的测试对象,因为它们的结合环可能变化很大且常缺乏强进化信号。通过先在大型通用数据集上训练ProTact,然后在抗体–抗原复合体上微调,作者展示了比其他专用工具更好的接触预测和对接质量。在一个针对SARS-CoV-2病毒的抗体案例研究中,即便序列信息有限,ProTact 仍生成了远比竞争方法更准确的复合体。这表明对表面进行详细的几何推理可以在一定程度上弥补缺乏进化线索的不足。
这一工作对未来的意义
对非专业读者来说,关键信息是ProTact 学会“感知”两种蛋白表面如何契合,而不是主要依赖长期的进化记录。通过将关于形状互补性的物理理念融入现代神经网络,它对蛋白质接触位置和如何组装成复合体提供了更可靠的估计。尽管它仍依赖于起始结构的质量,并且在蛋白质发生剧烈构象变化时表现欠佳,ProTact 为在数据稀缺环境中绘制蛋白配对图谱提供了强有力的新工具。这有望加速细胞机器机制的基础研究,并支持通过重塑蛋白相互作用来设计新疗法的工作。
引用: Rao, J., Liu, D., Zhou, X. et al. Accurate protein-protein interactions modeling through physics-informed geometric invariant learning. Commun Biol 9, 685 (2026). https://doi.org/10.1038/s42003-026-09809-2
关键词: 蛋白质-蛋白质相互作用, 结构预测, 对接, AlphaFold, 抗体结合