Clear Sky Science · es
Modelado preciso de interacciones proteína-proteína mediante aprendizaje geométrico invariante informado por la física
Por qué importan las asociaciones proteicas
Dentro de cada célula, las proteínas rara vez actúan de forma aislada. Se encuentran entre sí, encajan como piezas de un rompecabezas y forman asociaciones temporales que regulan señales, transportan cargas y combaten infecciones. Determinar con precisión cómo se encuentran dos proteínas en tres dimensiones puede revelar cómo surgen las enfermedades y cómo diseñar nuevos fármacos o anticuerpos. Pero obtener estas "instantáneas" moleculares en el laboratorio es lento y costoso, y las herramientas de IA actuales siguen fallando cuando los datos son escasos. Este estudio presenta ProTact, un nuevo método computacional que pretende cubrir esas carencias.
Límites de las herramientas actuales de predicción de estructuras
Sistemas como AlphaFold han transformado nuestra capacidad para predecir la forma de proteínas individuales y de algunos complejos. Funcionan mejor cuando hay muchas secuencias relacionadas disponibles, porque aprenden de patrones evolutivos. Sin embargo, muchos objetivos importantes, incluidos proteínas virales, proteínas diseñadas y anticuerpos, carecen de historiales evolutivos ricos. En esos casos, las herramientas existentes pueden equivocarse al juzgar cómo se ensamblan las proteínas, especialmente en los puntos de contacto precisos donde los átomos de dos socios se tocan. Los autores sostienen que, para superar esto, los modelos deben mirar más allá de los patrones de secuencia y prestar mayor atención a la forma física y al encaje de las superficies proteicas.
Una nueva forma de leer las superficies proteicas
ProTact trata cada proteína como una red de residuos conectados en el espacio y se centra en cómo sus superficies exteriores se complementan entre sí. Emplea un codificador geométrico diseñado cuidadosamente para que rotar o trasladar la proteína en el espacio no cambie la representación interna. Un segundo módulo aplica reglas inspiradas en la trigonometría para razonar sobre triángulos formados por grupos de residuos, ayudando al modelo a capturar efectos de muchos cuerpos en lugar de limitarse a contactos pareados. Juntas, estas partes generan un mapa que destaca qué residuos de las dos proteínas probablemente entren en contacto cuando se forma el complejo. ProTact puede trabajar con estructuras experimentales de alta calidad o con formas predichas por otras herramientas, y puede usar información de secuencia cuando esté disponible sin depender exclusivamente de ella.
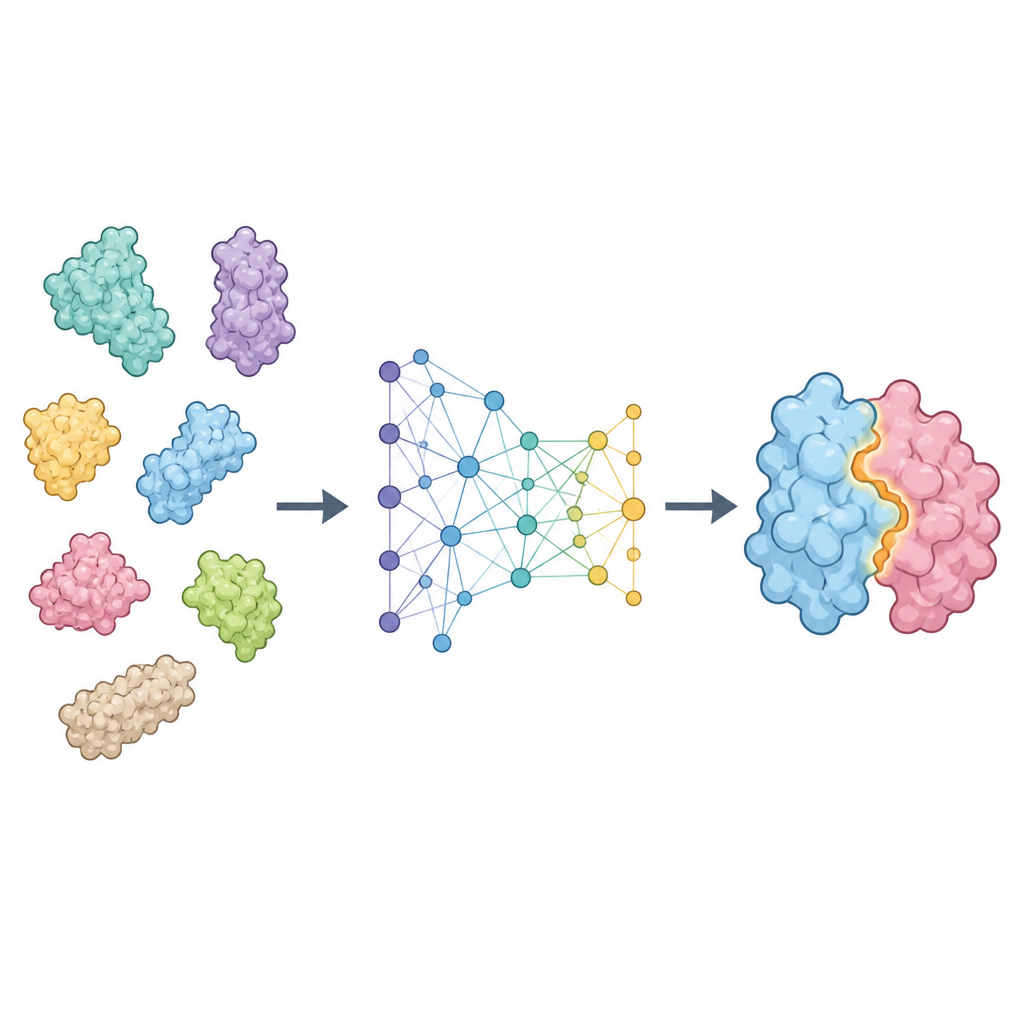
Qué tan bien funciona el método
Los investigadores probaron ProTact en varias colecciones estándar de pares de proteínas, incluidos casos desafiantes de experimentos de evaluación comunitaria. En todos estos puntos de referencia, ProTact identificó de forma consistente los residuos de contacto con mayor exactitud que los métodos líderes basados en secuencia y en estructura. Por ejemplo, mejoró una puntuación habitual de precisión en aproximadamente un tercio respecto al mejor enfoque existente en dos conjuntos de datos ampliamente utilizados. Incluso cuando las estructuras de entrada procedían de AlphaFold en lugar de experimentos, y por tanto contenían ruido, ProTact siguió siendo más preciso que las herramientas competidoras. El método también manejó una gran prueba ciega con pares de proteínas recientemente resueltos, mostrando un rendimiento sólido tanto para complejos simétricos como asimétricos.
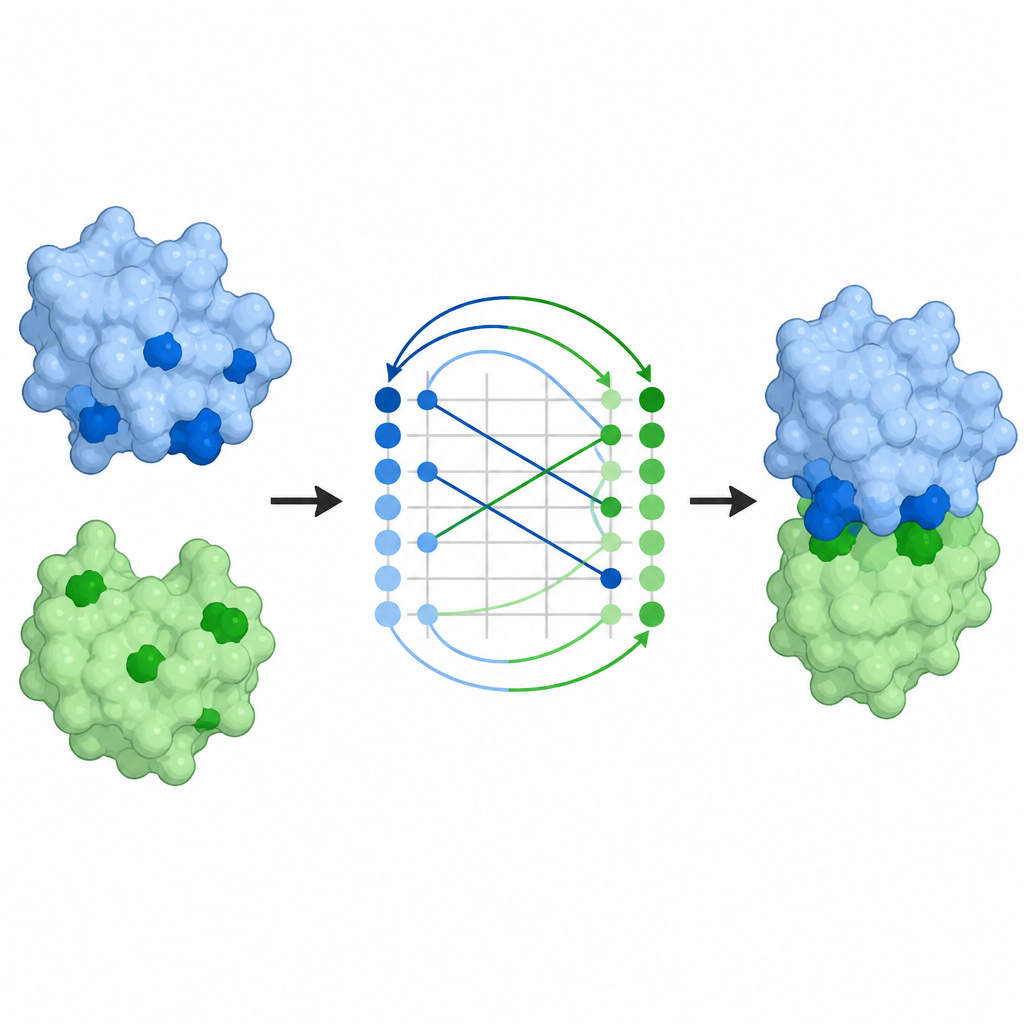
De mapas de contacto a complejos completos
Predecir qué residuos se tocan es solo parte del problema; los científicos también desean la disposición tridimensional completa de los socios. Los mapas de contacto de ProTact pueden convertirse en poses de docking, es decir, modelos completos de cómo se sitúan dos proteínas entre sí. Usando un algoritmo de alineamiento estándar, los autores muestran que las poses sugeridas por ProTact están más cercanas a los complejos determinados experimentalmente que las de otros métodos basados en contactos. Añadir estas predicciones de contacto como restricciones a programas de docking existentes mejora su precisión en la mayoría de los pares probados. ProTact también puede reordenar candidatos de complejos producidos por AlphaFold3, acomodándose mejor a las puntuaciones de calidad experimentales y mejorando los resultados en varios ejemplos de anticuerpo–antígeno donde los datos de secuencia son escasos.
Implicaciones para anticuerpos y diseño de fármacos
Los anticuerpos son una prueba particularmente difícil porque sus lazos de unión pueden variar ampliamente y con frecuencia carecen de señales evolutivas fuertes. Al entrenar primero a ProTact con un gran conjunto de datos general y después afinarlo con complejos anticuerpo–antígeno, los autores demuestran una mejor predicción de contactos y una mayor calidad de docking en comparación con otras herramientas especializadas. En un estudio de caso de un anticuerpo dirigido al virus SARS-CoV-2, ProTact produjo un complejo mucho más preciso que los métodos competidores, incluso con información de secuencia limitada. Esto sugiere que el razonamiento geométrico detallado sobre las superficies puede compensar en parte la falta de pistas evolutivas.
Qué significa este trabajo de cara al futuro
Para un no especialista, el mensaje clave es que ProTact aprende a "sentir" cómo encajan dos superficies proteicas, en lugar de apoyarse principalmente en amplios registros evolutivos. Al entretejer ideas físicas sobre la complementariedad de formas dentro de una red neuronal moderna, ofrece conjeturas más fiables sobre dónde se tocan las proteínas y cómo se ensamblan en complejos. Si bien todavía depende de la calidad de las estructuras de partida y tiene dificultades cuando las proteínas cambian de forma de manera drástica, ProTact ofrece una nueva herramienta potente para cartografiar asociaciones proteicas, especialmente en escenarios con pocos datos. Esto podría acelerar los estudios básicos de la maquinaria celular y apoyar el diseño de nuevos terapéuticos que actúen modulando interacciones proteicas.
Cita: Rao, J., Liu, D., Zhou, X. et al. Accurate protein-protein interactions modeling through physics-informed geometric invariant learning. Commun Biol 9, 685 (2026). https://doi.org/10.1038/s42003-026-09809-2
Palabras clave: interacciones proteína-proteína, predicción de estructuras, docking, AlphaFold, unión de anticuerpos