Clear Sky Science · it
Modelizzazione accurata delle interazioni proteina-proteina tramite apprendimento geometrico invariante informato dalla fisica
Perché le partnership proteiche sono importanti
All’interno di ogni cellula, le proteine raramente agiscono da sole. Si scontrano tra loro, si incastrano come pezzi di un puzzle e formano partnership temporanee che regolano segnali, trasportano carichi e combattono le infezioni. Capire esattamente come due proteine si incontrano in tre dimensioni può rivelare come insorgono le malattie e come progettare nuovi farmaci o anticorpi. Ma ottenere questi “istantanee” molecolari in laboratorio è lento e costoso, e gli strumenti di IA odierni faticano ancora quando i dati sono scarsi. Questo studio introduce ProTact, un nuovo metodo computazionale che mira a colmare queste lacune.
Limiti degli attuali strumenti di predizione strutturale
Sistemi come AlphaFold hanno trasformato la nostra capacità di prevedere la forma di singole proteine e di alcuni complessi. Funzionano meglio quando sono disponibili molte sequenze correlate, perché apprendono da schemi evolutivi. Tuttavia, molti bersagli importanti, inclusi proteine virali, proteine ingegnerizzate e anticorpi, non hanno ricche storie evolutive. In questi casi, gli strumenti esistenti possono valutare erroneamente come le proteine si assemblano, specialmente nei punti di contatto precisi in cui gli atomi di due partner si toccano. Gli autori sostengono che per superare questo limite i modelli devono guardare oltre i pattern di sequenza e prestare maggiore attenzione alla forma fisica e all’accoppiamento delle superfici proteiche.
Un nuovo modo di leggere le superfici proteiche
ProTact tratta ogni proteina come una rete di residui connessi nello spazio e si concentra su come le loro superfici esterne si completano reciprocamente. Utilizza un codificatore geometrico progettato in modo che la rotazione o la traslazione della proteina nello spazio non alteri la rappresentazione interna. Un secondo modulo applica regole ispirate alla trigonometria per ragionare sui triangoli formati da gruppi di residui, aiutando il modello a catturare effetti multi-corpo invece dei soli contatti coppia-per-coppia. Insieme, queste componenti producono una mappa che evidenzia quali residui dei due partner sono probabilmente a contatto quando si forma il complesso. ProTact può funzionare con strutture sperimentali di alta qualità o con forme predette da altri strumenti, e può usare informazioni di sequenza quando disponibili senza dipenderne esclusivamente.
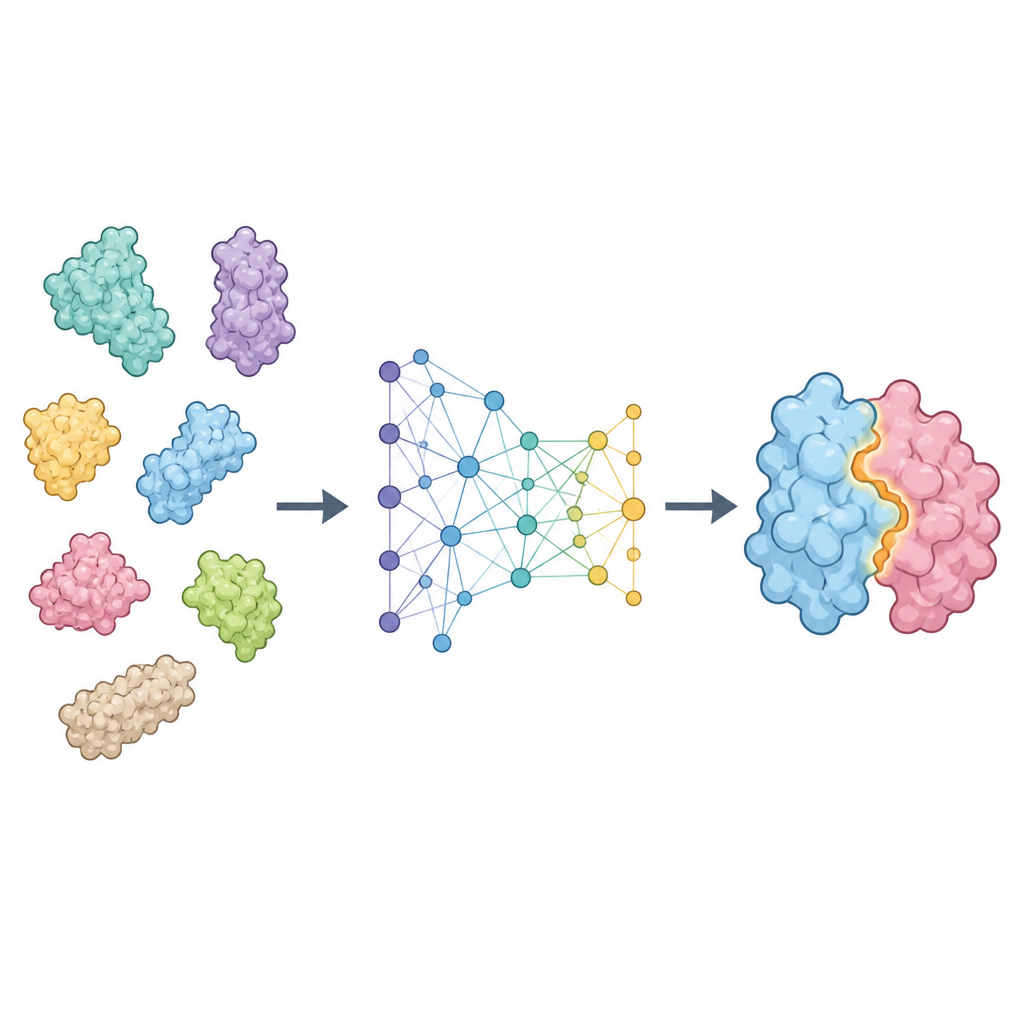
Quanto bene funziona il metodo
I ricercatori hanno testato ProTact su diverse collezioni standard di coppie proteiche, inclusi casi difficili provenienti da esperimenti di valutazione a livello comunitario. In questi benchmark, ProTact ha identificato in modo più accurato i residui di contatto rispetto ai principali metodi basati su sequenza e su struttura. Per esempio, ha migliorato un punteggio di precisione comune di circa un terzo rispetto al miglior approccio esistente su due dataset ampiamente usati. Anche quando le strutture in ingresso provenivano da AlphaFold anziché da dati sperimentali, e quindi contenevano rumore, ProTact è rimasto più accurato rispetto agli strumenti concorrenti. Il metodo ha inoltre gestito un ampio test cieco di coppie proteiche recentemente risolte, mostrando prestazioni solide sia per complessi simmetrici che asimmetrici.
Dalle mappe di contatto ai complessi completi
Predire quali residui si toccano è solo una parte del problema; gli scienziati vogliono anche l’intero arrangiamento tridimensionale dei partner. Le mappe di contatto di ProTact possono essere convertite in pose di docking, cioè modelli completi di come due proteine si dispongono insieme. Utilizzando un algoritmo di allineamento standard, gli autori mostrano che le pose suggerite da ProTact sono più vicine ai complessi determinati sperimentalmente rispetto a quelle prodotte da altri metodi basati sui contatti. Aggiungere queste previsioni di contatto come vincoli ai programmi di docking esistenti migliora la loro accuratezza sulla maggior parte delle coppie testate. ProTact può anche riallineare le complessità candidate prodotte da AlphaFold3, aderendo più strettamente ai punteggi di qualità sperimentali e migliorando i risultati per diversi esempi di antigene–anticorpo dove i dati di sequenza sono scarsi.
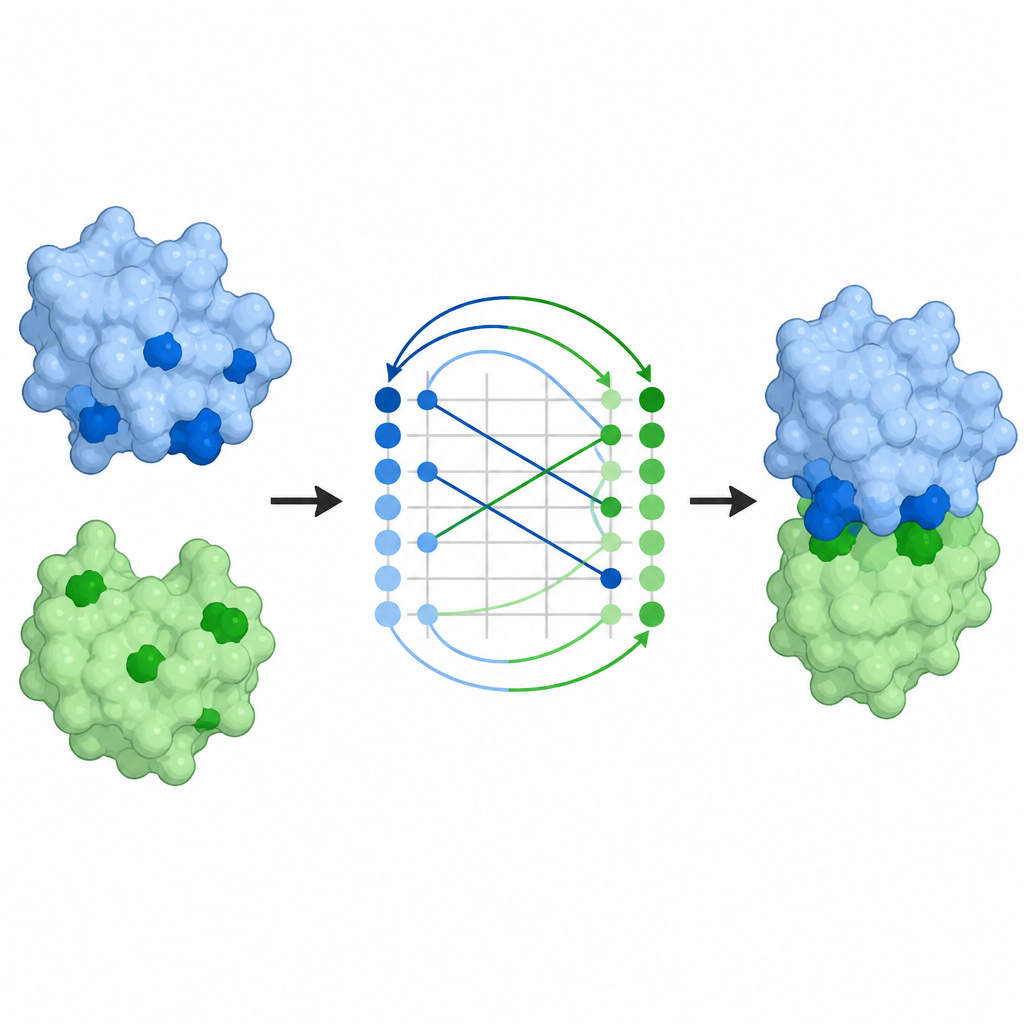
Implicazioni per anticorpi e progettazione di farmaci
Gli anticorpi sono un test particolarmente difficile perché i loro anelli di legame possono variare molto e spesso mancano di forti segnali evolutivi. Addestrando prima ProTact su un ampio dataset generale e poi affinandolo su complessi antigene–anticorpo, gli autori mostrano un miglioramento nella predizione dei contatti e nella qualità del docking rispetto ad altri strumenti specializzati. In uno studio di caso su un anticorpo che prende di mira il virus SARS-CoV-2, ProTact ha prodotto un complesso molto più accurato rispetto ai metodi concorrenti, anche con informazioni di sequenza limitate. Questo suggerisce che un ragionamento geometrico dettagliato sulle superfici può compensare in parte la mancanza di indizi evolutivi.
Cosa significa questo lavoro per il futuro
Per un non specialista, il messaggio chiave è che ProTact impara a “percepire” come due superfici proteiche si incastrano, invece di affidarsi principalmente a lunghi record evolutivi. Intrecciando concetti fisici di complementarità di forma in una rete neurale moderna, fornisce ipotesi più affidabili su dove le proteine si toccano e su come si assemblano in complessi. Pur dipendendo ancora dalla qualità delle strutture iniziali e incontrando difficoltà quando le proteine cambiano forma in modo drammatico, ProTact offre un nuovo strumento potente per mappare le partnership proteiche, specialmente in contesti con pochi dati. Questo potrebbe accelerare studi di base sulla macchina cellulare e supportare la progettazione di nuovi terapeutici che agiscono rimodellando le interazioni proteiche.
Citazione: Rao, J., Liu, D., Zhou, X. et al. Accurate protein-protein interactions modeling through physics-informed geometric invariant learning. Commun Biol 9, 685 (2026). https://doi.org/10.1038/s42003-026-09809-2
Parole chiave: interazioni proteina-proteina, predizione della struttura, docking, AlphaFold, legame degli anticorpi