Clear Sky Science · de
Genaue Modellierung von Protein‑Protein‑Interaktionen durch physikinformiertes geometrisch invariantes Lernen
Warum Proteinpartnerschaften wichtig sind
Innerhalb jeder Zelle arbeiten Proteine selten allein. Sie stoßen aneinander, fügen sich wie Puzzleteile zusammen und bilden temporäre Partnerschaften, die Signale steuern, Fracht transportieren und Infektionen bekämpfen. Zu verstehen, wie zwei Proteine sich dreidimensional treffen, kann aufdecken, wie Krankheiten entstehen und wie man neue Wirkstoffe oder Antikörper entwirft. Solche molekularen „Momentaufnahmen“ im Labor zu erstellen ist jedoch zeitaufwendig und teuer, und heutige KI‑Werkzeuge geraten ins Stolpern, wenn Daten knapp sind. Diese Studie führt ProTact ein, eine neue rechnerische Methode, die diese Lücken zu schließen versucht.
Grenzen aktueller Strukturvorhersage‑Tools
Systeme wie AlphaFold haben unsere Fähigkeit, die Form einzelner Proteine und einiger Komplexe vorherzusagen, grundlegend verändert. Sie funktionieren am besten, wenn viele verwandte Proteinsequenzen verfügbar sind, da sie aus evolutiven Mustern lernen. Viele wichtige Ziele – etwa virale Proteine, gentechnisch veränderte Proteine und Antikörper – verfügen jedoch nicht über reichhaltige evolutionäre Historien. In solchen Fällen können bestehende Werkzeuge fehlinterpretieren, wie Proteine zusammenkommen, insbesondere an den präzisen Kontaktpunkten, an denen Atome der beiden Partner aneinanderstoßen. Die Autoren argumentieren, dass Modelle hierfür über sequenzielle Muster hinausblicken und der physischen Form und Passung der Proteinoberflächen größere Aufmerksamkeit schenken müssen.
Eine neue Methode, Proteinoberflächen zu lesen
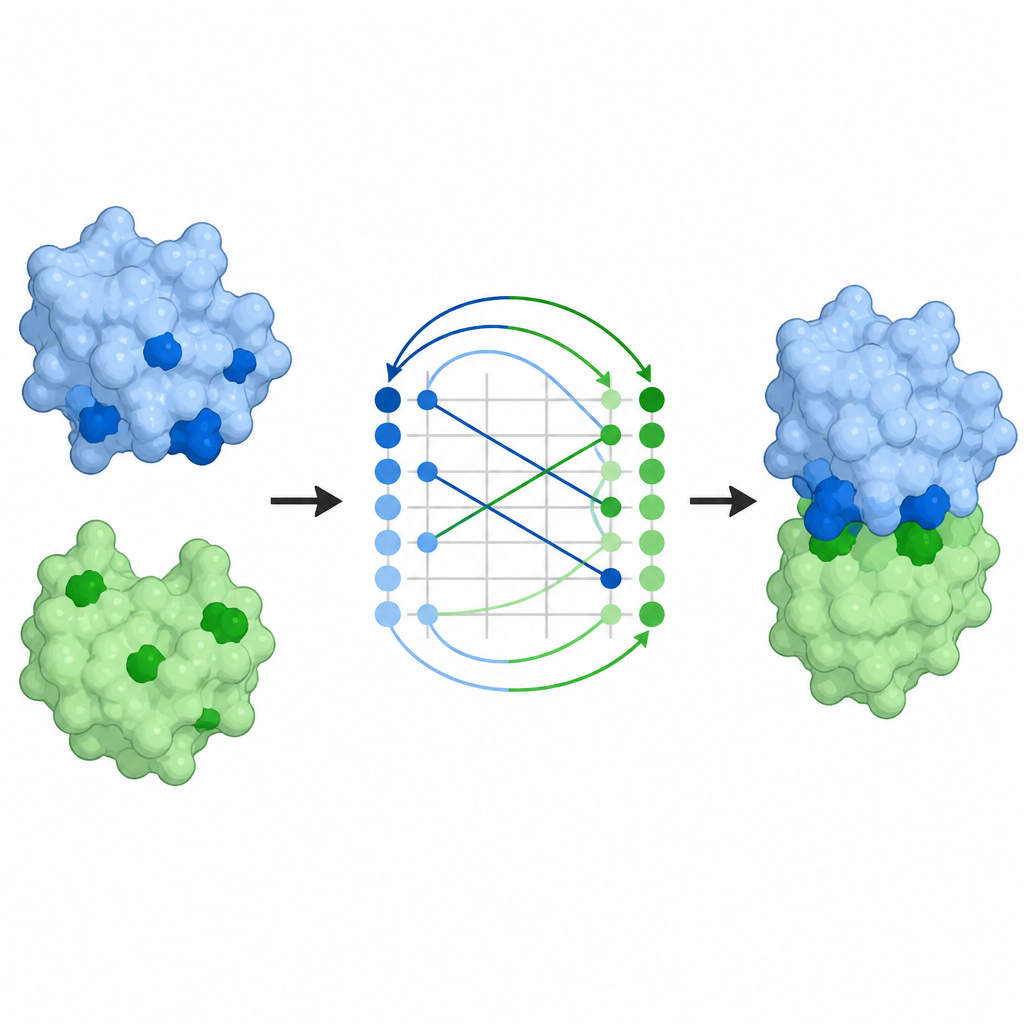
ProTact behandelt jedes Protein als Netzwerk von in Raum verbundenen Resten und konzentriert sich darauf, wie deren äußere Oberflächen zueinander komplementär sind. Es verwendet einen geometrischen Encoder, der so konstruiert ist, dass Drehungen oder Verschiebungen des Proteins im Raum die interne Darstellung nicht verändern. Ein zweites Modul wendet trigonometrisch inspirierte Regeln an, um über Dreiecke zu schließen, die durch Gruppen von Resten gebildet werden, und hilft dem Modell, Viele‑Körper‑Effekte statt nur paarweiser Kontakte zu erfassen. Zusammen erzeugen diese Komponenten eine Karte, die hervorhebt, welche Reste der beiden Proteine wahrscheinlich in Kontakt stehen, wenn der Komplex gebildet wird. ProTact kann mit hochwertigen experimentellen Strukturen arbeiten oder mit von anderen Werkzeugen vorhergesagten Formen, und es kann Sequenzinformationen nutzen, wenn sie verfügbar sind, ohne von ihnen abhängig zu sein.
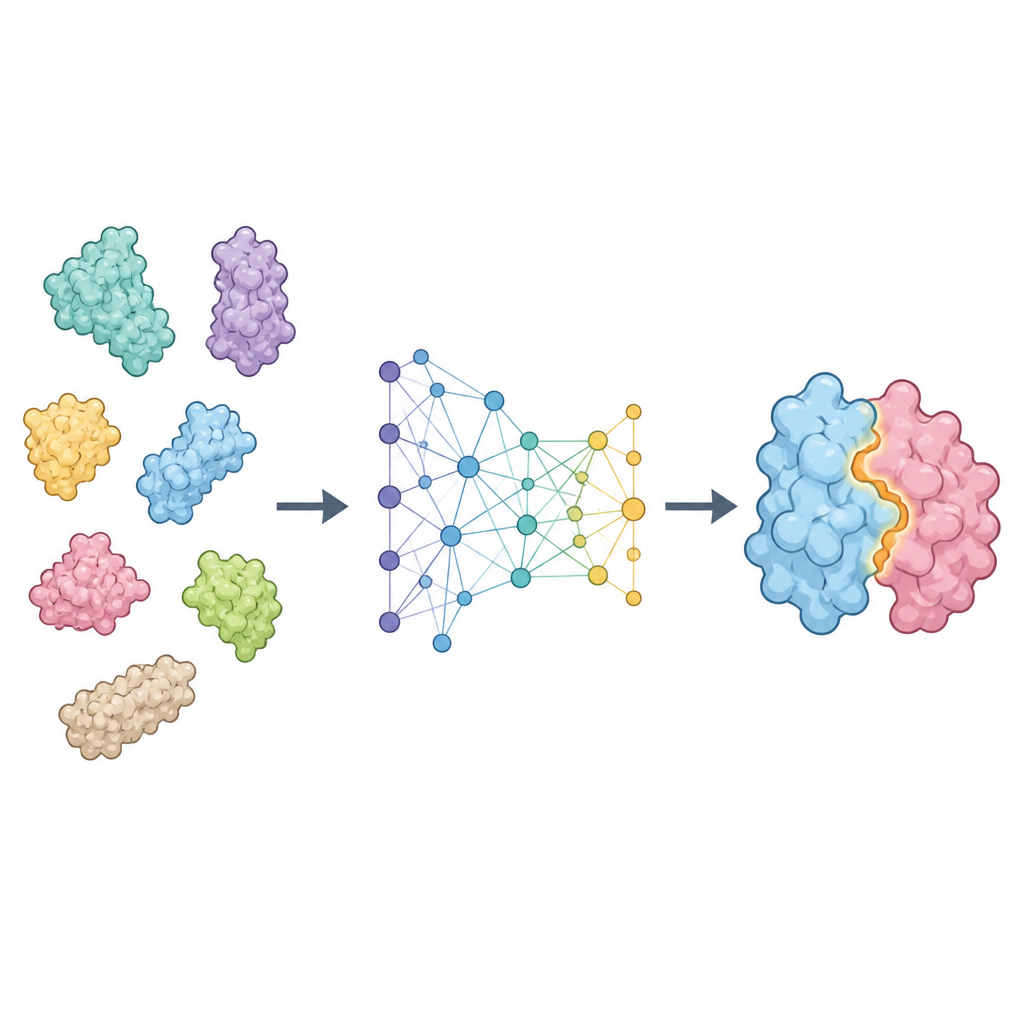
Wie gut die Methode abschneidet
Die Forscher testeten ProTact an mehreren standardisierten Sammlungen von Proteinpaaren, einschließlich herausfordernder Fälle aus gemeinschaftlichen Bewertungs‑Experimenten. Über diese Benchmarks hinweg identifizierte ProTact Kontaktreste konsistent genauer als führende sequenzbasierte und strukturbasierte Methoden. So verbesserte es beispielsweise einen gängigen Präzisionswert um rund ein Drittel im Vergleich zur besten vorhandenen Methode bei zwei weit verbreiteten Datensätzen. Selbst wenn die Eingangsstrukturen von AlphaFold statt aus Experimenten stammten und daher Rauschen enthielten, blieb ProTact genauer als konkurrierende Werkzeuge. Die Methode meisterte auch einen großen Blindtest mit kürzlich gelösten Proteinpaaren und zeigte starke Leistung sowohl für symmetrische als auch asymmetrische Komplexe.
Von Kontaktkarten zu vollständigen Komplexen
Die Vorhersage, welche Reste sich berühren, ist nur ein Teil des Problems; Wissenschaftler wollen auch die vollständige dreidimensionale Anordnung der Partner. ProTacts Kontaktkarten lassen sich in Docking‑Posen umwandeln, also in vollständige Modelle, wie zwei Proteine zueinander sitzen. Mit einem Standard‑Ausrichtungsalgorithmus zeigen die Autoren, dass ProTacts vorgeschlagene Posen näher an experimentell bestimmten Komplexen liegen als die anderer kontaktbasierter Methoden. Das Hinzufügen dieser Kontaktvorhersagen als Restriktionen zu bestehenden Docking‑Programmen verbessert deren Genauigkeit bei den meisten getesteten Paaren. ProTact kann zudem Kandidatenkomplexe, die von AlphaFold3 erzeugt wurden, neu bewerten, experimentelle Qualitätswerte besser treffen und die Ergebnisse für mehrere Antikörper–Antigen‑Beispiele verbessern, in denen Sequenzdaten spärlich sind.
Implikationen für Antikörper und Wirkstoffdesign
Antikörper sind ein besonders schwieriger Prüfstand, weil ihre Bindeschleifen stark variieren können und oft keine starken evolutionären Signale aufweisen. Durch das Vortrainieren von ProTact auf einem großen allgemeinen Datensatz und anschließendes Feinabstimmen auf Antikörper–Antigen‑Komplexe zeigen die Autoren verbesserte Kontaktvorhersage und Docking‑Qualität im Vergleich zu anderen spezialisierten Werkzeugen. In einer Fallstudie zu einem Antikörper gegen das SARS‑CoV‑2‑Virus erzeugte ProTact einen deutlich genaueren Komplex als konkurrierende Methoden, selbst bei begrenzten Sequenzinformationen. Das deutet darauf hin, dass detaillierte geometrische Überlegungen zur Oberflächenkomplementarität fehlende evolutionäre Hinweise teilweise ausgleichen können.
Was diese Arbeit für die Zukunft bedeutet
Für Nicht‑Spezialisten lautet die Kernbotschaft: ProTact lernt, „zu erspüren“, wie zwei Proteinoberflächen zusammenpassen, anstatt sich hauptsächlich auf lange evolutionäre Sequenzen zu stützen. Indem physikalische Ideen zur Formkomplementarität in ein modernes neuronales Netz eingeflochten werden, liefert es verlässlichere Vermutungen darüber, wo Proteine sich berühren und wie sie zu Komplexen zusammensetzen. Obwohl die Methode weiterhin von der Qualität der Ausgangsstrukturen abhängt und Probleme hat, wenn Proteine ihre Form dramatisch ändern, bietet ProTact ein kraftvolles neues Werkzeug zum Kartieren von Proteinpartnerschaften, insbesondere in datenarmen Situationen. Das könnte grundlegende Studien der zellulären Maschinerie beschleunigen und das Design neuer Therapeutika unterstützen, die durch Umgestaltung von Proteininteraktionen wirken.
Zitation: Rao, J., Liu, D., Zhou, X. et al. Accurate protein-protein interactions modeling through physics-informed geometric invariant learning. Commun Biol 9, 685 (2026). https://doi.org/10.1038/s42003-026-09809-2
Schlüsselwörter: Protein‑Protein‑Interaktionen, Strukturvorhersage, Docking, AlphaFold, Antikörperbindung