Clear Sky Science · sv
Noggrann modellering av protein–proteininteraktioner genom fysikinformerad geometriskt invariant inlärning
Varför proteinpartnerskap är viktiga
Inuti varje cell arbetar proteiner sällan ensamma. De stöter på varandra, hakar i varandra som pusselbitar och bildar tillfälliga partnerskap som styr signaler, transporterar last och bekämpar infektioner. Att fastställa exakt hur två proteiner möts i tre dimensioner kan avslöja hur sjukdomar uppstår och hur man designar nya läkemedel eller antikroppar. Men att ta fram dessa molekylära »ögonblicksbilder« i laboratoriet är långsamt och dyrt, och dagens AI-verktyg snubblar fortfarande när data är knapphändiga. Denna studie introducerar ProTact, en ny beräkningsmetod som syftar till att fylla dessa luckor.
Begränsningar hos nuvarande strukturprediktionsverktyg
System som AlphaFold har omvandlat vår förmåga att förutsäga formen hos enskilda proteiner och vissa komplex. De fungerar bäst när många besläktade proteinsekvenser finns tillgängliga, eftersom de lär sig från evolutionära mönster. Många viktiga mål, inklusive virala proteiner, designade proteiner och antikroppar, saknar dock rika evolutionära historiespår. I dessa fall kan befintliga verktyg felbedöma hur proteiner går ihop, särskilt vid de precisa kontaktpunkter där atomer från två partners vidrör varandra. Författarna menar att för att övervinna detta måste modeller se bortom sekvensmönster och uppmärksamma den fysiska formen och passformen hos proteinytor.
Ett nytt sätt att läsa proteinytor
ProTact behandlar varje protein som ett nätverk av rester kopplade i rummet och fokuserar på hur deras yttre ytor kompletterar varandra. Den använder en geometrisk enkodare som är noggrant utformad så att rotation eller förskjutning av proteinet i rummet inte ändrar den interna representationen. En andra modul tillämpar trigonometriskt inspirerade regler för att resonera om trianglar bildade av grupper av rester, vilket hjälper modellen att fånga många-kropps-effekter snarare än enbart parvisa kontakter. Tillsammans ger dessa delar en karta som lyfter fram vilka rester från de två proteinerna som sannolikt kommer att vara i kontakt när komplexet bildas. ProTact kan arbeta med högkvalitativa experimentella strukturer eller med former som förutsagts av andra verktyg, och kan använda sekvensinformation när den finns utan att vara beroende av den.
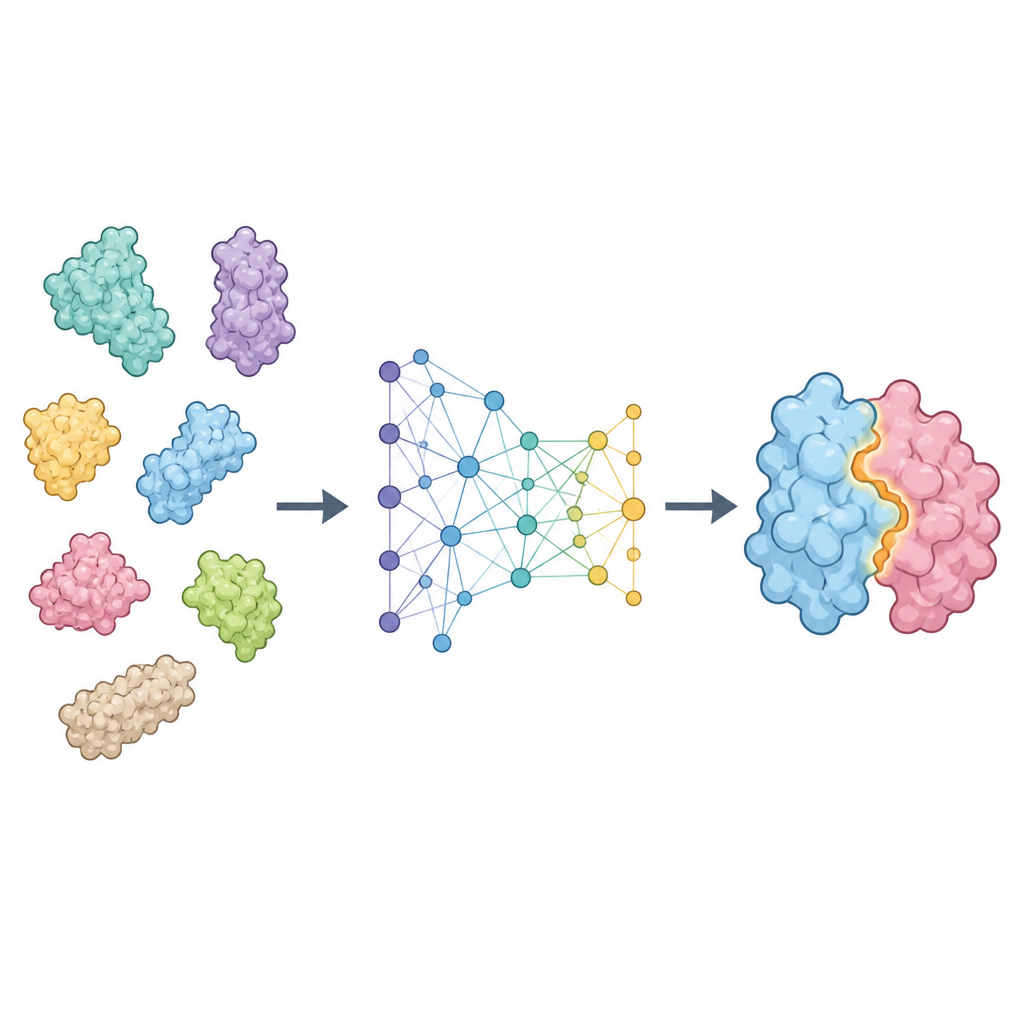
Hur väl metoden presterar
Forskarna testade ProTact på flera standardkollektioner av proteinpar, inklusive svåra fall från community-omfattande utvärderingsexperiment. Över dessa benchmarkar identifierade ProTact konsekvent kontaktrester mer korrekt än ledande metod baserade på sekvens och struktur. Till exempel förbättrade den en vanlig precisionpoäng med ungefär en tredjedel jämfört med den bästa befintliga metoden på två vida använda dataset. Även när indata-strukturerna kom från AlphaFold snarare än experiment och därför innehöll brus, förblev ProTact mer exakt än konkurrerande verktyg. Metoden hanterade också ett stort blint test av nyligen lösta proteinpar och visade stark prestanda för både symmetriska och asymmetriska komplex.
Från kontaktkartor till fullständiga komplex
Att förutsäga vilka rester som vidrör varandra är bara en del av problemet; forskare vill också ha den fullständiga tredimensionella arrangemanget av partnerna. ProTacts kontaktkartor kan omvandlas till dockningspositioner, det vill säga fullständiga modeller av hur två proteiner sitter ihop. Med en standardiserad aligneringsalgoritm visar författarna att ProTacts föreslagna positioner ligger närmare experimentellt bestämda komplex än de från andra kontaktbaserade metoder. Att lägga till dessa kontaktprediktioner som restriktioner i befintliga dockningsprogram förbättrar deras noggrannhet för de flesta testade par. ProTact kan också omrankera kandidatkomplex som producerats av AlphaFold3, vilket bättre överensstämmer med experimentella kvalitetsmått och förbättrar resultat för flera antikropp–antigen-exempel där sekvensdata är knappa.
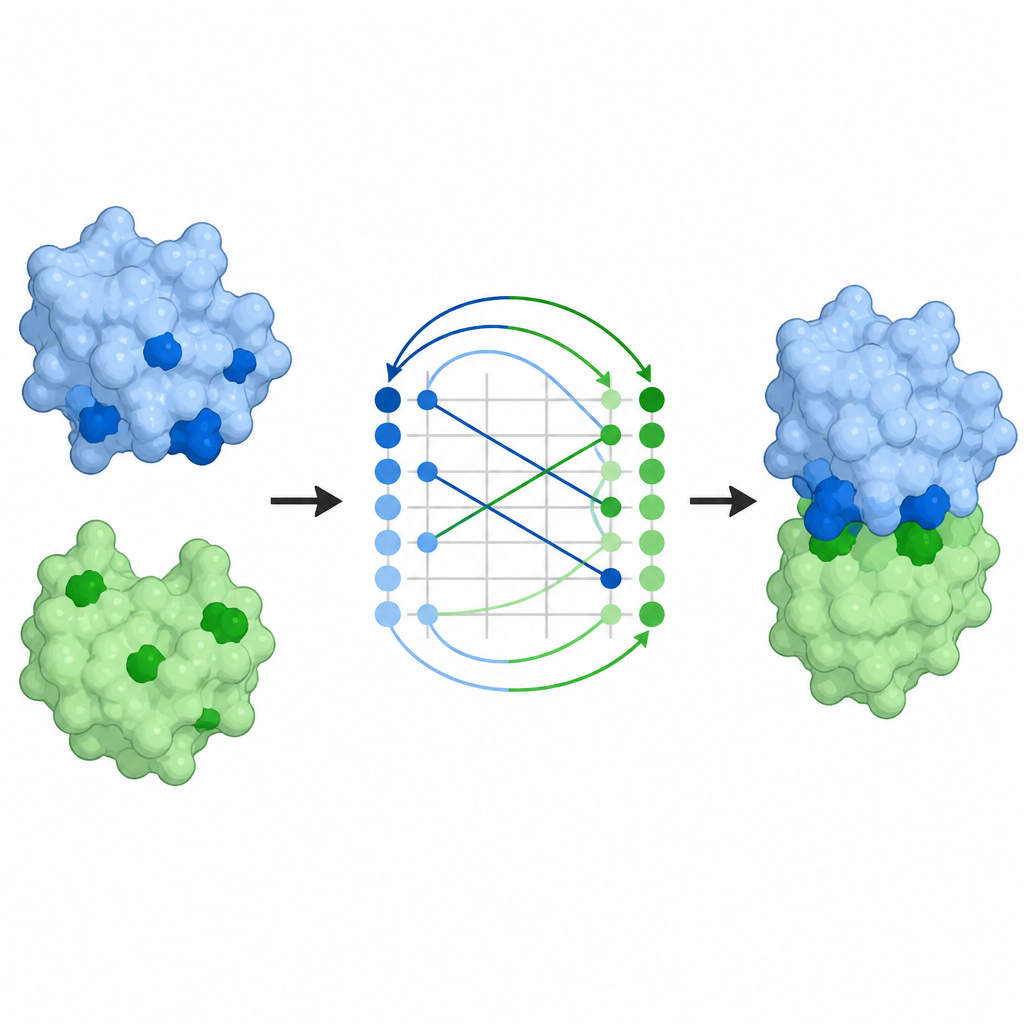
Konsekvenser för antikroppar och läkemedelsdesign
Antikroppar är ett särskilt svårt test eftersom deras bindningsslingor kan variera kraftigt och ofta saknar starka evolutionära signaler. Genom att först träna ProTact på ett stort generellt dataset och sedan finjustera det på antikropp–antigen-komplex visar författarna förbättrad kontaktprediktion och dockningskvalitet jämfört med andra specialiserade verktyg. I en fallstudie av en antikropp som riktar sig mot SARS-CoV-2 producerade ProTact ett mycket mer korrekt komplex än konkurrerande metoder, även med begränsad sekvensinformation. Detta antyder att detaljerat geometriskt resonerande om ytor delvis kan kompensera för bristen på evolutionära ledtrådar.
Vad detta arbete innebär framöver
För en icke-specialist är huvudbudskapet att ProTact lär sig att »känna efter« hur två proteinytor passar ihop, snarare än att i första hand förlita sig på långa evolutionära register. Genom att väva in fysiska idéer om formkomplementaritet i ett modernt neuralt nätverk ger det mer pålitliga gissningar om var proteiner vidrör varandra och hur de sätter ihop sig till komplex. Även om det fortfarande beror på kvaliteten hos startstrukturerna och har svårigheter när proteiner ändrar form dramatiskt, erbjuder ProTact ett kraftfullt nytt verktyg för att kartlägga proteinpartnerskap, särskilt i datafattiga miljöer. Detta kan snabba upp grundläggande studier av cellens maskineri och stödja designen av nya terapier som fungerar genom att omforma proteininteraktioner.
Citering: Rao, J., Liu, D., Zhou, X. et al. Accurate protein-protein interactions modeling through physics-informed geometric invariant learning. Commun Biol 9, 685 (2026). https://doi.org/10.1038/s42003-026-09809-2
Nyckelord: protein–proteininteraktioner, strukturprediktion, dockning, AlphaFold, antikroppsbindning