Clear Sky Science · fr
Modélisation précise des interactions protéine–protéine via un apprentissage géométrique invariant informé par la physique
Pourquoi les partenariats protéiques comptent
À l’intérieur de chaque cellule, les protéines travaillent rarement isolément. Elles se rencontrent, s’emboîtent comme des pièces de puzzle et forment des associations temporaires qui contrôlent les signaux, transportent des cargos et combattent les infections. Déterminer précisément comment deux protéines se rencontrent en trois dimensions peut révéler les origines de maladies et guider la conception de nouveaux médicaments ou anticorps. Mais obtenir ces « instantanés » moléculaires en laboratoire est lent et coûteux, et les outils d’IA actuels pataugent encore quand les données sont rares. Cette étude introduit ProTact, une nouvelle méthode computationnelle destinée à combler ces lacunes.
Limites des outils actuels de prédiction de structure
Des systèmes comme AlphaFold ont transformé notre capacité à prédire la forme de protéines isolées et de certains complexes. Ils fonctionnent mieux lorsqu’un grand nombre de séquences apparentées est disponible, car ils apprennent à partir de motifs évolutifs. Cependant, de nombreuses cibles importantes, y compris des protéines virales, des protéines conçues et des anticorps, manquent d’histoires évolutives riches. Dans ces cas, les outils existants peuvent mal évaluer la façon dont les protéines s’assemblent, en particulier aux points de contact précis où des atomes de deux partenaires se touchent. Les auteurs soutiennent que, pour surmonter cela, les modèles doivent regarder au-delà des motifs de séquence et prêter une attention plus étroite à la forme physique et à l’ajustement des surfaces protéiques.
Une nouvelle façon d’analyser les surfaces protéiques
ProTact considère chaque protéine comme un réseau de résidus connectés dans l’espace et se concentre sur la complémentarité de leurs surfaces externes. Il utilise un encodeur géométrique conçu pour que la rotation ou la translation de la protéine dans l’espace n’altère pas la représentation interne. Un second module applique des règles inspirées de la trigonométrie pour raisonner sur les triangles formés par des groupes de résidus, aidant le modèle à capter des effets à plusieurs corps plutôt que de se limiter aux contacts par paires. Ensemble, ces composants produisent une carte qui met en évidence quels résidus des deux protéines sont susceptibles d’être en contact lors de la formation du complexe. ProTact peut fonctionner avec des structures expérimentales de haute qualité ou avec des formes prédites par d’autres outils, et il peut utiliser l’information de séquence quand elle est disponible sans en dépendre exclusivement.
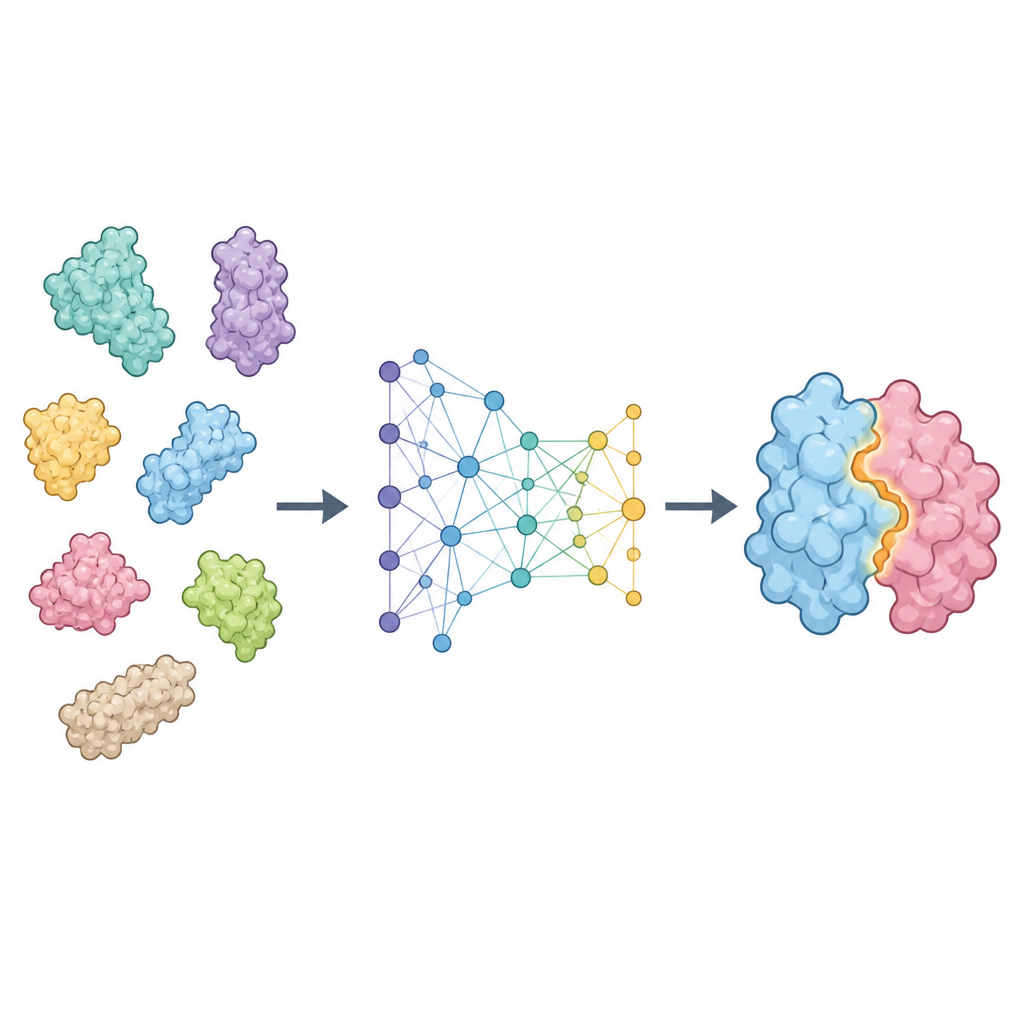
Performance de la méthode
Les chercheurs ont testé ProTact sur plusieurs jeux de paires protéiques standard, y compris des cas difficiles issus d’expériences d’évaluation communautaire. Sur ces benchmarks, ProTact a systématiquement identifié les résidus de contact plus précisément que les méthodes de référence basées sur la séquence ou la structure. Par exemple, il a amélioré un score de précision courant d’environ un tiers par rapport à la meilleure approche existante sur deux ensembles de données largement utilisés. Même lorsque les structures d’entrée provenaient d’AlphaFold plutôt que d’expériences, et contenaient donc du bruit, ProTact est resté plus précis que les outils concurrents. La méthode a aussi bien géré un large test à l’aveugle de paires protéiques récemment résolues, montrant de bonnes performances pour des complexes symétriques et asymétriques.
Des cartes de contact aux complexes complets
Prédire quels résidus se touchent ne résout qu’une partie du problème ; les scientifiques veulent aussi l’arrangement tridimensionnel complet des partenaires. Les cartes de contact de ProTact peuvent être converties en poses de docking, c’est-à-dire en modèles complets de l’assemblage de deux protéines. En utilisant un algorithme d’alignement standard, les auteurs montrent que les poses suggérées par ProTact sont plus proches des complexes déterminés expérimentalement que celles issues d’autres méthodes basées sur les contacts. Ajouter ces prédictions de contact comme contraintes aux programmes de docking existants améliore leur précision sur la plupart des paires testées. ProTact peut également reranker des complexes candidats produits par AlphaFold3, se rapprochant davantage des scores expérimentaux de qualité et améliorant les résultats pour plusieurs exemples anticorps–antigène lorsque les données de séquence sont rares.
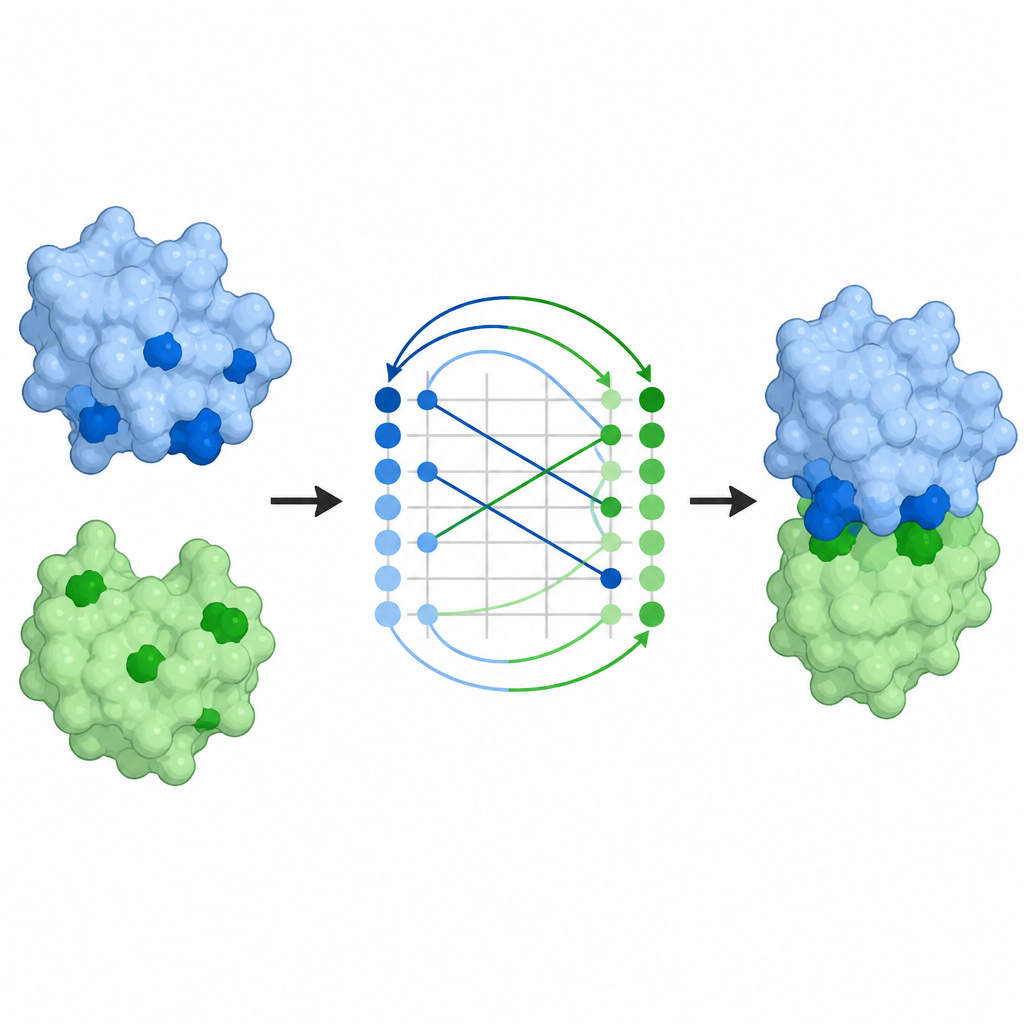
Implications pour les anticorps et la conception de médicaments
Les anticorps constituent un test particulièrement difficile car leurs boucles de liaison peuvent varier fortement et manquent souvent de signaux évolutifs marqués. En entraînant d’abord ProTact sur un large jeu de données général, puis en l’affinant sur des complexes anticorps–antigène, les auteurs montrent une amélioration de la prédiction des contacts et de la qualité du docking par rapport à d’autres outils spécialisés. Dans une étude de cas portant sur un anticorps ciblant le SARS‑CoV‑2, ProTact a produit un complexe bien plus précis que les méthodes concurrentes, même avec des informations de séquence limitées. Cela suggère que le raisonnement géométrique détaillé sur les surfaces peut compenser en partie l’absence d’indices évolutifs.
Ce que ce travail implique pour l’avenir
Pour un non-spécialiste, le message clé est que ProTact apprend à « ressentir » comment deux surfaces protéiques s’emboîtent, plutôt que de dépendre principalement de longs enregistrements évolutifs. En intégrant des idées physiques sur la complémentarité de forme dans un réseau neuronal moderne, il fournit des estimations plus fiables sur les points de contact et la manière dont les protéines s’assemblent en complexes. Bien qu’il dépende encore de la qualité des structures de départ et peine lorsque les protéines changent de conformation de manière dramatique, ProTact offre un nouvel outil puissant pour cartographier les partenariats protéiques, notamment dans les contextes pauvres en données. Cela pourrait accélérer les études fondamentales du fonctionnement cellulaire et soutenir la conception de nouveaux thérapeutiques visant à remodeler les interactions protéiques.
Citation: Rao, J., Liu, D., Zhou, X. et al. Accurate protein-protein interactions modeling through physics-informed geometric invariant learning. Commun Biol 9, 685 (2026). https://doi.org/10.1038/s42003-026-09809-2
Mots-clés: interactions protéine–protéine, prédiction de structure, docking, AlphaFold, liaison des anticorps