Clear Sky Science · en
Accurate protein-protein interactions modeling through physics-informed geometric invariant learning
Why protein partnerships matter
Inside every cell, proteins rarely work alone. They bump into each other, lock together like puzzle pieces, and form temporary partnerships that control signals, carry cargo, and fight infections. Working out exactly how two proteins meet in three dimensions can reveal how diseases arise and how to design new drugs or antibodies. But making these molecular "snapshots" in the lab is slow and expensive, and today’s AI tools still stumble when data are scarce. This study introduces ProTact, a new computational method that aims to fill those gaps.
Limits of current structure prediction tools
Systems such as AlphaFold have transformed our ability to predict the shape of single proteins and some complexes. They work best when many related protein sequences are available, because they learn from patterns of evolution. However, many important targets, including viral proteins, engineered proteins, and antibodies, lack rich evolutionary histories. In these cases, existing tools can misjudge how proteins come together, especially at the precise contact points where atoms from two partners touch. The authors argue that to overcome this, models must look beyond sequence patterns and pay closer attention to the physical shape and fit of protein surfaces.
A new way to read protein surfaces
ProTact treats each protein as a network of residues connected in space and focuses on how their outer surfaces complement each other. It uses a geometric encoder that is carefully designed so that rotating or shifting the protein in space does not change the internal representation. A second module applies trigonometry-inspired rules to reason about triangles formed by groups of residues, helping the model capture many-body effects rather than just pairwise contacts. Together, these parts produce a map that highlights which residues from the two proteins are likely to be in contact when the complex forms. ProTact can work with high-quality experimental structures or with shapes predicted by other tools, and it can use sequence information when available without depending on it.
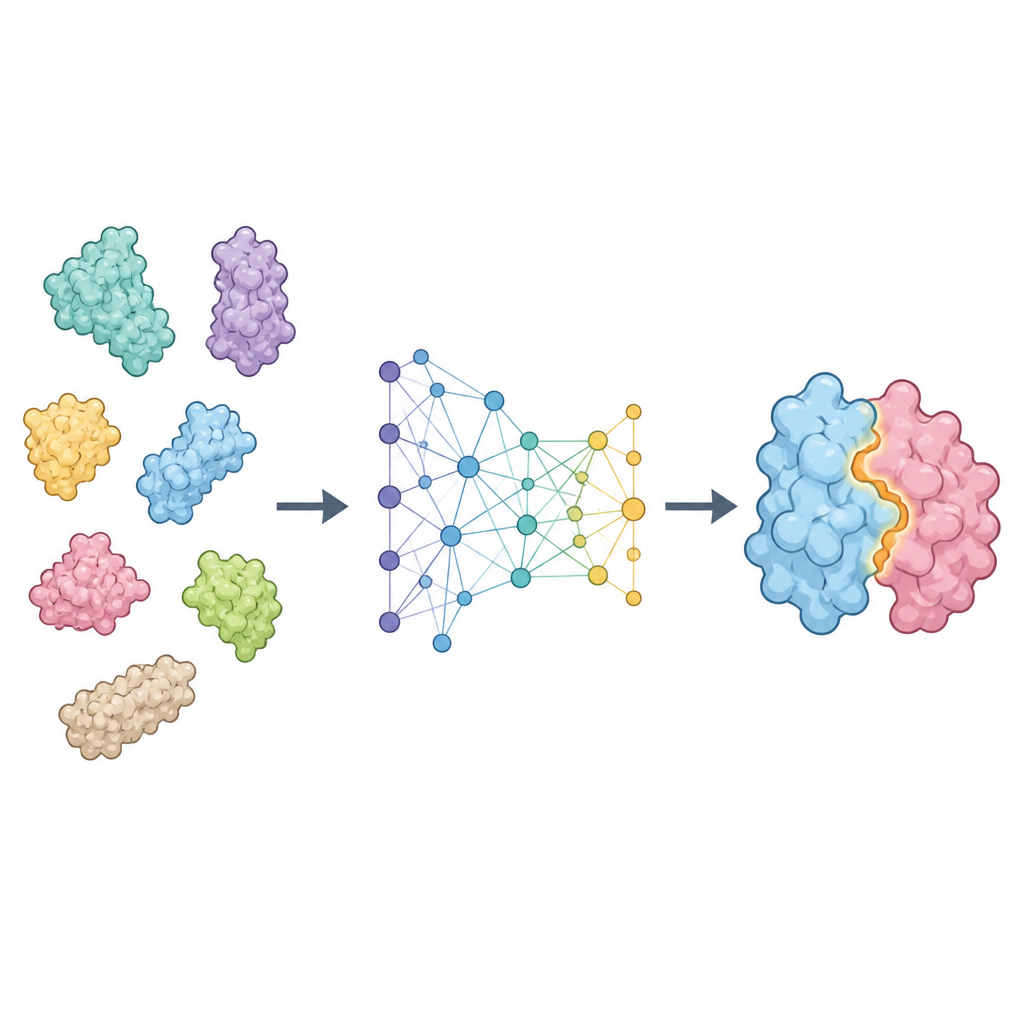
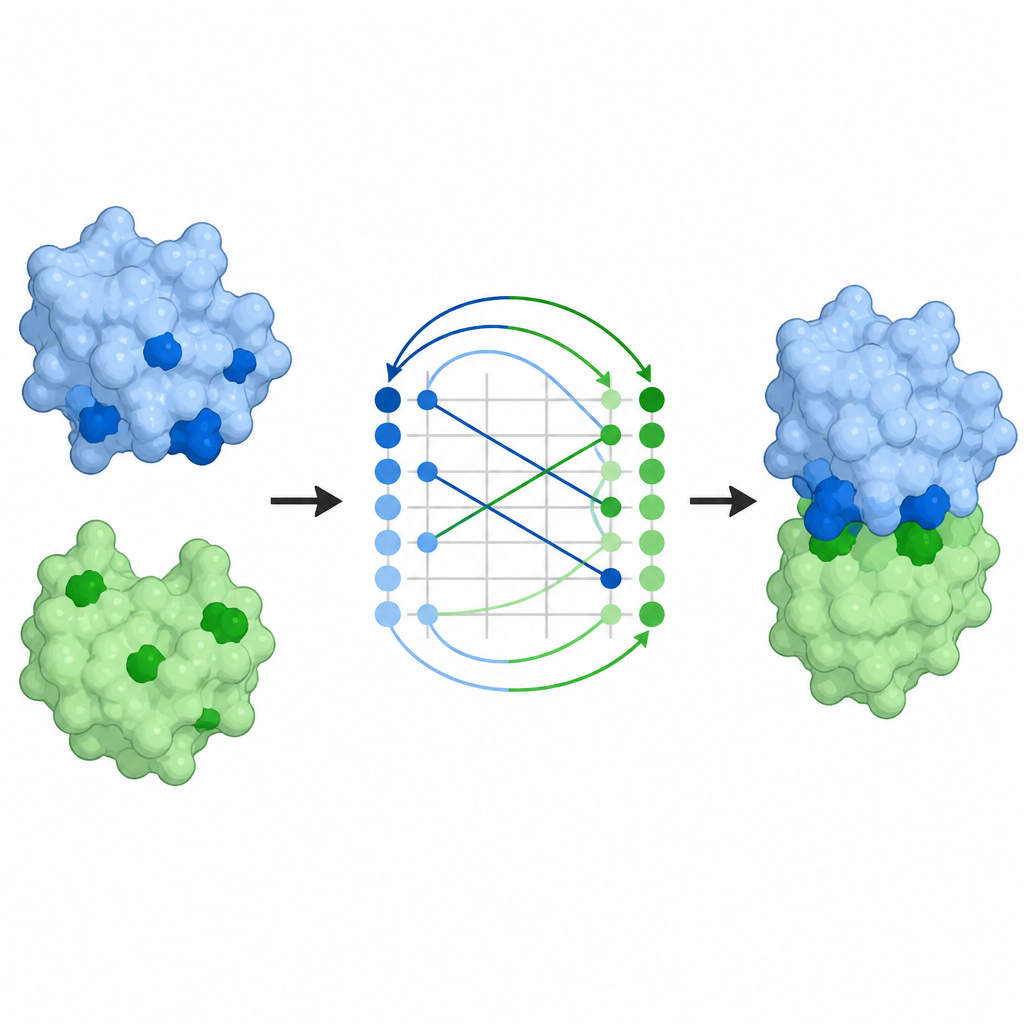
How well the method performs
The researchers tested ProTact on several standard collections of protein pairs, including challenging cases from community-wide assessment experiments. Across these benchmarks, ProTact consistently identified contact residues more accurately than leading sequence-based and structure-based methods. For example, it improved a common precision score by around one-third compared with the best existing approach on two widely used datasets. Even when the input structures came from AlphaFold rather than experiments, and therefore contained noise, ProTact remained more accurate than competing tools. The method also handled a large blind test of recently solved protein pairs, showing strong performance for both symmetric and asymmetric complexes.
From contact maps to full complexes
Predicting which residues touch is only part of the problem; scientists also want the full three-dimensional arrangement of the partners. ProTact’s contact maps can be converted into docking poses, that is, full models of how two proteins sit together. Using a standard alignment algorithm, the authors show that ProTact’s suggested poses are closer to experimentally determined complexes than those from other contact-based methods. Adding these contact predictions as restraints to existing docking programs improves their accuracy on most tested pairs. ProTact can also re-rank candidate complexes produced by AlphaFold3, more closely matching experimental quality scores and improving results for several antibody–antigen examples where sequence data are sparse.
Implications for antibodies and drug design
Antibodies are a particularly difficult test because their binding loops can vary widely and often lack strong evolutionary signals. By first training ProTact on a large general dataset and then fine-tuning it on antibody–antigen complexes, the authors show improved contact prediction and docking quality compared with other specialized tools. In a case study of an antibody targeting the SARS-CoV-2 virus, ProTact produced a much more accurate complex than competing methods, even with limited sequence information. This suggests that detailed geometric reasoning about surfaces can partly offset the lack of evolutionary clues.
What this work means going forward
To a non-specialist, the key message is that ProTact learns to "feel" how two protein surfaces fit together, rather than relying mainly on long evolutionary records. By weaving physical ideas about shape complementarity into a modern neural network, it provides more reliable guesses about where proteins touch and how they assemble into complexes. While it still depends on the quality of the starting structures and struggles when proteins change shape dramatically, ProTact offers a powerful new tool for mapping protein partnerships, especially in data-poor settings. This could speed up basic studies of cellular machinery and support the design of new therapeutics that work by reshaping protein interactions.
Citation: Rao, J., Liu, D., Zhou, X. et al. Accurate protein-protein interactions modeling through physics-informed geometric invariant learning. Commun Biol 9, 685 (2026). https://doi.org/10.1038/s42003-026-09809-2
Keywords: protein-protein interactions, structure prediction, docking, AlphaFold, antibody binding