Clear Sky Science · zh
SIRT1通过诱导FOXO1/自噬/NCOA4介导的铁死亡并抑制E2F1/NOTCH‑1/YAP信号通路,加速脑梗死的进展
这项卒中研究为何重要
脑梗死作为常见的卒中类型,仍然是全球范围内导致死亡和致残的主要原因之一。医生有时可以重新开放被阻塞的血管,但许多患者仍会留下持久的脑损伤。本研究深入细胞层面,揭示一种关键调控蛋白SIRT1如何通过推动细胞走向一种由铁驱动的毁灭性死亡形式来加重卒中后的损伤。理解这一隐蔽通路可能为在每一分钟都至关重要的情况下保护大脑开辟新的治疗途径。
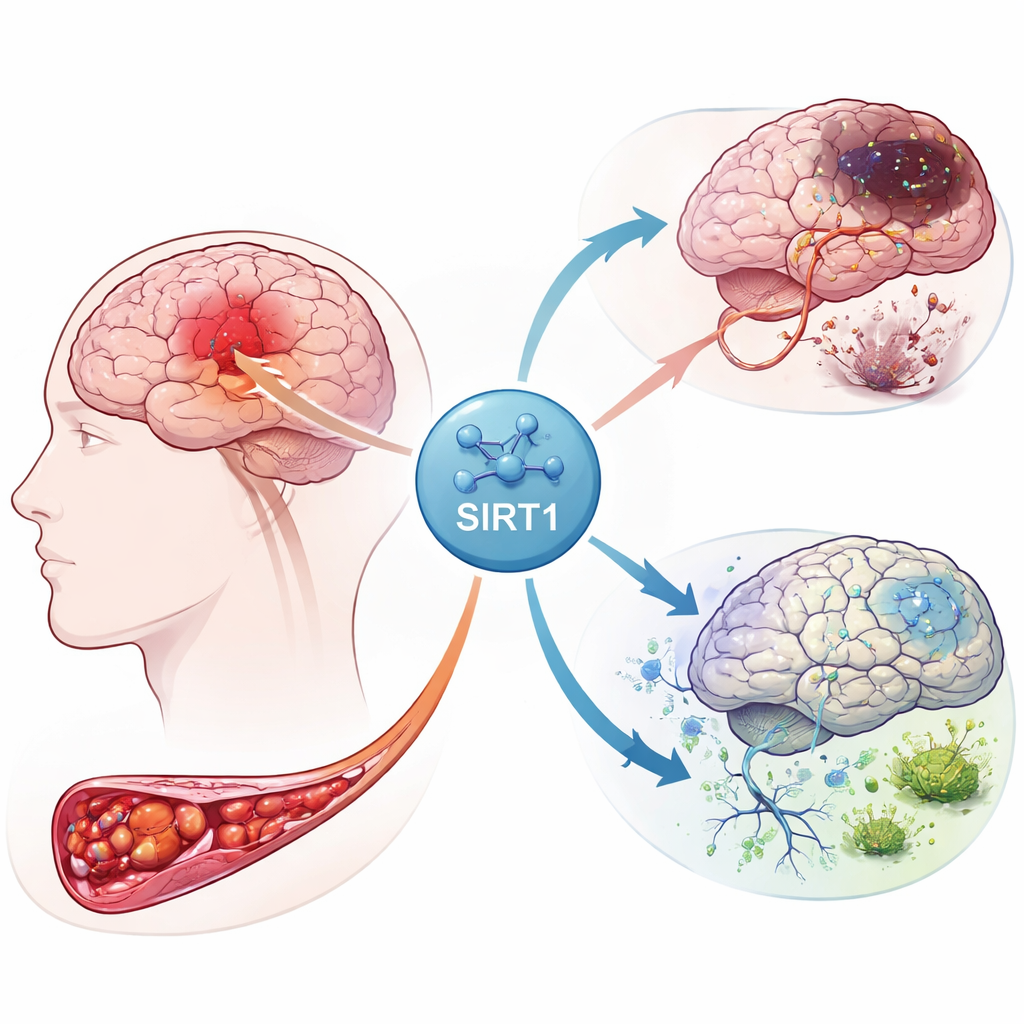
受损大脑内的连锁反应
当部分脑组织的血流被切断时,神经细胞会缺氧缺能量。这一危机在包括星形胶质细胞等多种细胞类型内引发信号风暴。作者把注意力放在名为SIRT1的蛋白上,该蛋白作为能量应激的主调节因子。利用来自卒中患者的基因表达数据,他们发现SIRT1与参与铁代谢、细胞存活以及一种新识别的细胞死亡形式——铁死亡有关基因密切相关。分析还凸显了SIRT1与数条已知调控细胞生长和自我清理过程的信号通路之间的联系。
从大数据到动物脑和细胞培养
为了解这些线索在活体组织中的表现,研究团队建立了大鼠脑梗死模型:短暂阻断一条主要脑动脉然后恢复血流。有些大鼠接受能激活SIRT1的药物处理,另一些则被注入能增强另一路径组分Notch‑1的病毒。脑切片显示卒中后SIRT1活性急剧上升,且上调SIRT1会使坏死组织面积增大并加重神经元损伤。相反,提高Notch‑1水平能减轻损伤。蛋白质检测证实了这一模式:当SIRT1升高时,与Notch‑1及其伴侣蛋白YAP相关的保护因子下降,而卒中损伤扩大。
铁与“自噬自食”如何驱动细胞死亡
研究者随后将实验移至暴露于低氧、低养分并添加铁的培养星形胶质细胞,以模拟卒中条件。通过对SIRT1、E2F1、Notch‑1、YAP和铁死亡的抑制剂或促动剂进行精确处理,他们绘制出一条详细的事件链。SIRT1升高会抑制细胞核中的E2F1、Notch‑1和YAP,但会上调另一调节因子FOXO1,同时增加标志自噬活性蛋白和名为NCOA4的货运适配蛋白。这一组合促成了一种特殊的自噬形式,能破坏铁储存复合物,导致细胞内自由铁激增。与此同时,抗氧化防御标志发生改变,细胞死亡率飙升,与铁死亡的生化特征相符。阻断铁死亡或恢复Notch‑1/YAP轴的部分环节可以减少这些有害变化。
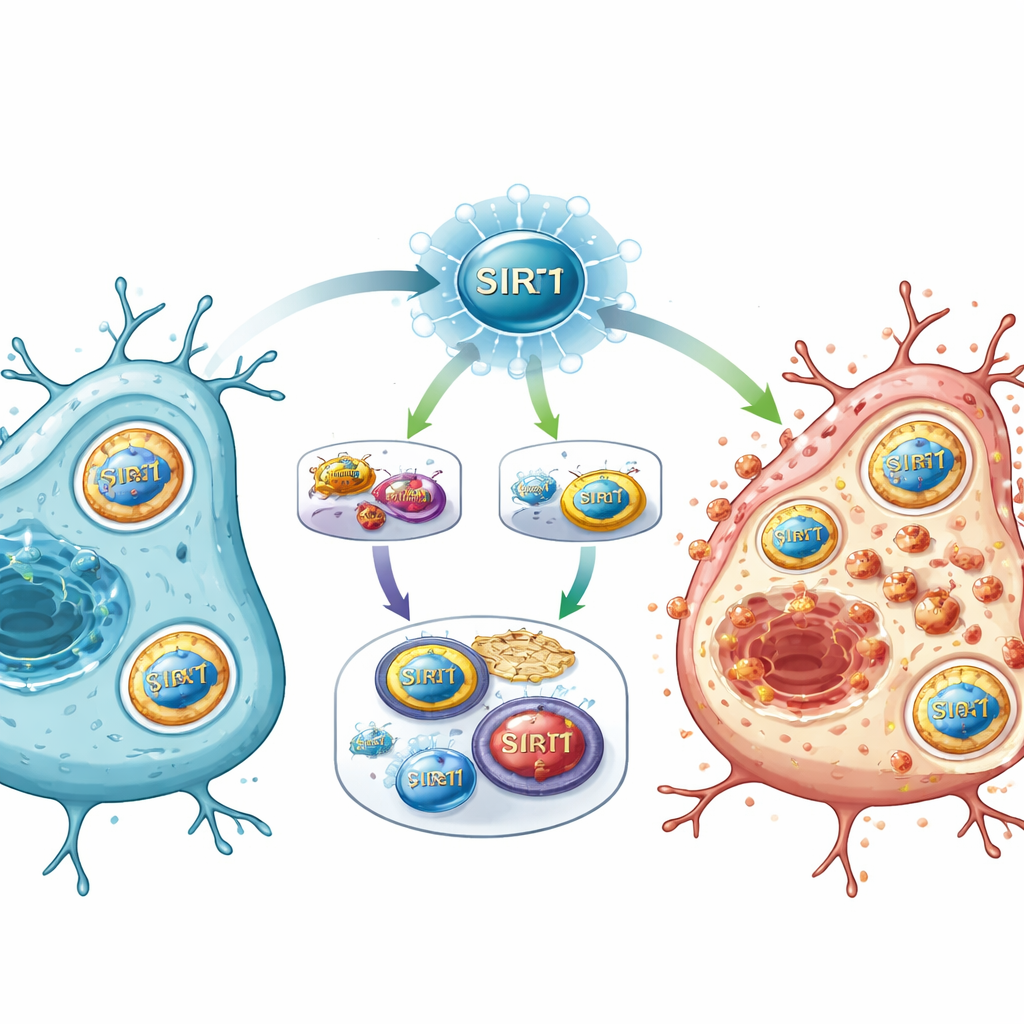
受伤通路的新“主开关”
综合这些发现,研究提出SIRT1在卒中后充当一个主开关,处于能量感知、基因调控和铁代谢的交汇点。当SIRT1被强烈激活时,它抑制通常有助于维持某些生存基因和抗氧化系统的E2F1/Notch‑1/YAP通路。与此同时,SIRT1又促进FOXO1及招募NCOA4拆解铁储存蛋白的自噬程序。由此产生的铁超载,加上化学防御能力减弱,会驱动细胞膜脂质损伤,并将细胞推向铁死亡。
这对未来卒中治疗意味着什么
对非专业读者来说,关键信息是卒中后的细胞死亡并非单一,其部分类型可能是可防的。这项工作表明,在受压迫的大脑中,SIRT1可以将平衡倾向于一种破坏性的铁依赖性死亡通路而非保护。针对SIRT1本身,或针对Notch‑1、YAP、FOXO1、自噬和NCOA4等下游步骤,可能提供超越单纯再通血管的新策略来限制脑损伤。尽管仍需在人体组织中开展更多研究,但本文勾勒的通路为设计能够抑制细胞内这场由铁驱动的“火”的药物并有望改善脑梗死后的恢复提供了路线图。
引用: Wang, G., Ma, S., Qi, L. et al. SIRT1 induces FOXO1/autophagy/NCOA4-induced ferroptosis and accelerates the progression of cerebral infarction by inhibiting the E2F1/NOTCH-1/YAP signaling pathway. Sci Rep 16, 13946 (2026). https://doi.org/10.1038/s41598-026-44355-1
关键词: 脑梗死, 铁死亡, SIRT1, 自噬, 卒中机制