Clear Sky Science · fr
SIRT1 induit la ferroptose via FOXO1/autophagie/NCOA4 et accélère la progression de l'infarctus cérébral en inhibant la voie de signalisation E2F1/NOTCH-1/YAP
Pourquoi cette étude sur l'AVC est importante
L'infarctus cérébral, une forme courante d'AVC, reste une des principales causes de mortalité et d'invalidité dans le monde. Les médecins peuvent parfois rouvrir des vaisseaux obstrués, mais de nombreux patients gardent des lésions cérébrales durables. Cette étude examine l'intérieur des cellules cérébrales pour découvrir comment une protéine de régulation clé, SIRT1, pourrait en réalité aggraver les lésions post-AVC en poussant les cellules vers une forme destructrice de mort cellulaire dépendante du fer. Comprendre cette voie cachée pourrait ouvrir la voie à de nouveaux traitements protégeant le cerveau lorsque chaque minute compte.
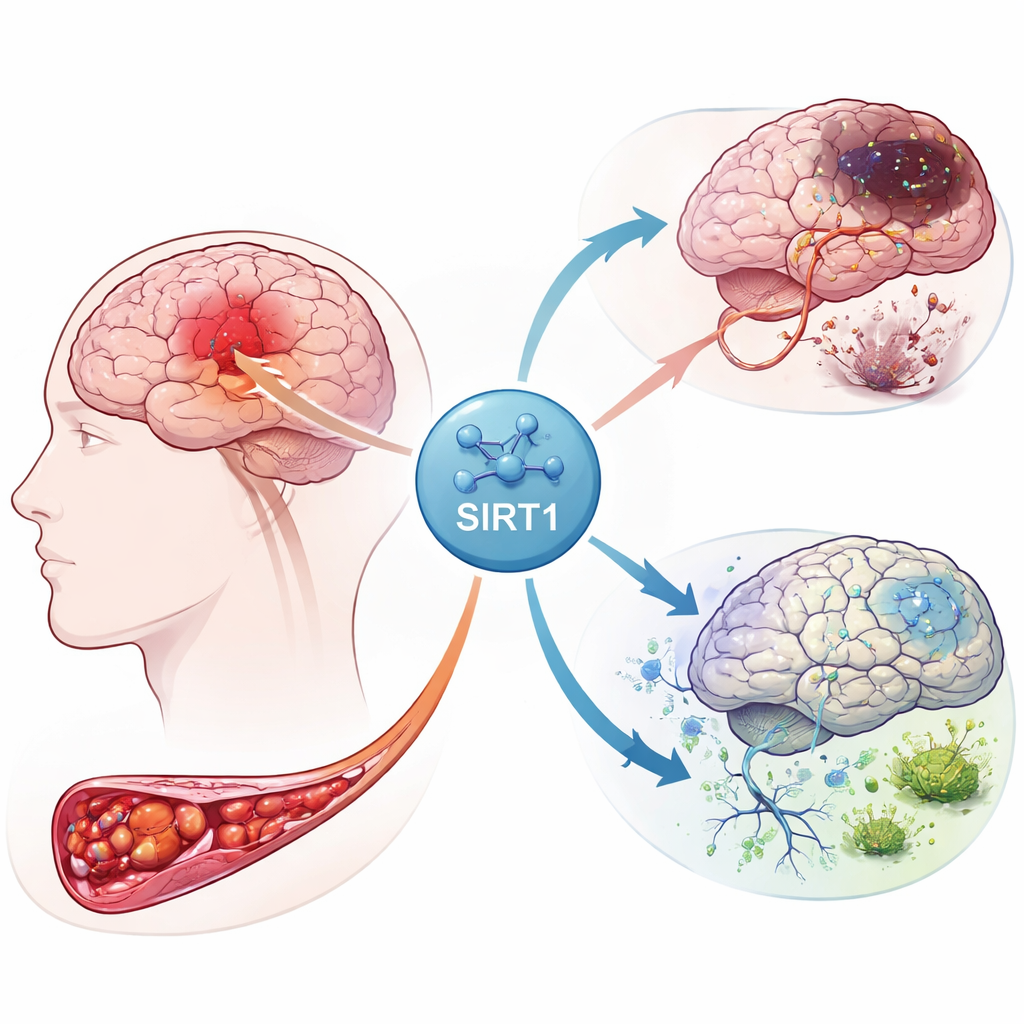
La réaction en chaîne à l'intérieur d'un cerveau endommagé
Lorsque l'apport sanguin à une partie du cerveau est interrompu, les neurones sont privés d'oxygène et d'énergie. Cette crise déclenche une tempête de signaux au sein de nombreux types cellulaires, y compris des cellules de soutien appelées astrocytes. Les auteurs se sont concentrés sur une protéine nommée SIRT1, un régulateur maître qui réagit au stress énergétique. En utilisant des données d'expression génique provenant de patients victimes d'AVC, ils ont découvert que SIRT1 était étroitement lié à des gènes impliqués dans le métabolisme du fer, la survie cellulaire et une forme de mort cellulaire récemment reconnue, la ferroptose. Leur analyse a également mis en évidence des connexions entre SIRT1 et plusieurs voies de signalisation déjà connues pour contrôler la croissance cellulaire et les processus d'auto-nettoyage.
Des mégadonnées aux cerveaux d'animaux et aux cultures cellulaires
Pour voir comment ces indices se manifestent dans les tissus vivants, l'équipe a créé un modèle de rat d'infarctus cérébral en bloquant brièvement une artère cérébrale majeure puis en rétablissant le flux sanguin. Certains rats ont reçu un médicament activant SIRT1, tandis que d'autres ont été traités par un virus augmentant un autre composant de la voie, Notch-1. Des coupes cérébrales ont montré que l'activité de SIRT1 augmentait fortement après l'AVC et que l'augmentation de SIRT1 agrandissait la zone de tissu nécrosé et amplifiait les lésions neuronales. En revanche, l'élévation des niveaux de Notch-1 réduisait les dommages. Les dosages protéiques ont confirmé ce schéma : lorsque SIRT1 était élevé, les facteurs protecteurs associés à Notch-1 et à une protéine partenaire, YAP, diminuaient, tandis que les lésions post-AVC s'étendaient.
Comment le fer et l'autophagie conduisent à la mort cellulaire
Les chercheurs sont ensuite passés à des astrocytes en culture exposés à un faible niveau d'oxygène, à une faible disponibilité en nutriments et à un apport de fer pour imiter les conditions de l'AVC. En ajoutant avec précaution des inhibiteurs ou des activateurs de SIRT1, E2F1, Notch-1, YAP et de la ferroptose, ils ont cartographié une chaîne d'événements détaillée. Un SIRT1 élevé réduisait l'activité nucléaire d'E2F1, Notch-1 et YAP, mais augmentait un autre régulateur appelé FOXO1, ainsi que des protéines marquant une autophagie accrue et un adaptateur de cargaison nommé NCOA4. Cette combinaison favorisait un type particulier d'autophagie qui démantèle les complexes de stockage du fer, inondant les cellules de fer libre. Pendant ce processus, les marqueurs des défenses antioxydantes se modifiaient et la mortalité cellulaire augmentait fortement, correspondant au profil biochimique de la ferroptose. Bloquer la ferroptose ou restaurer des éléments de l'axe Notch-1/YAP réduisait ces changements délétères.
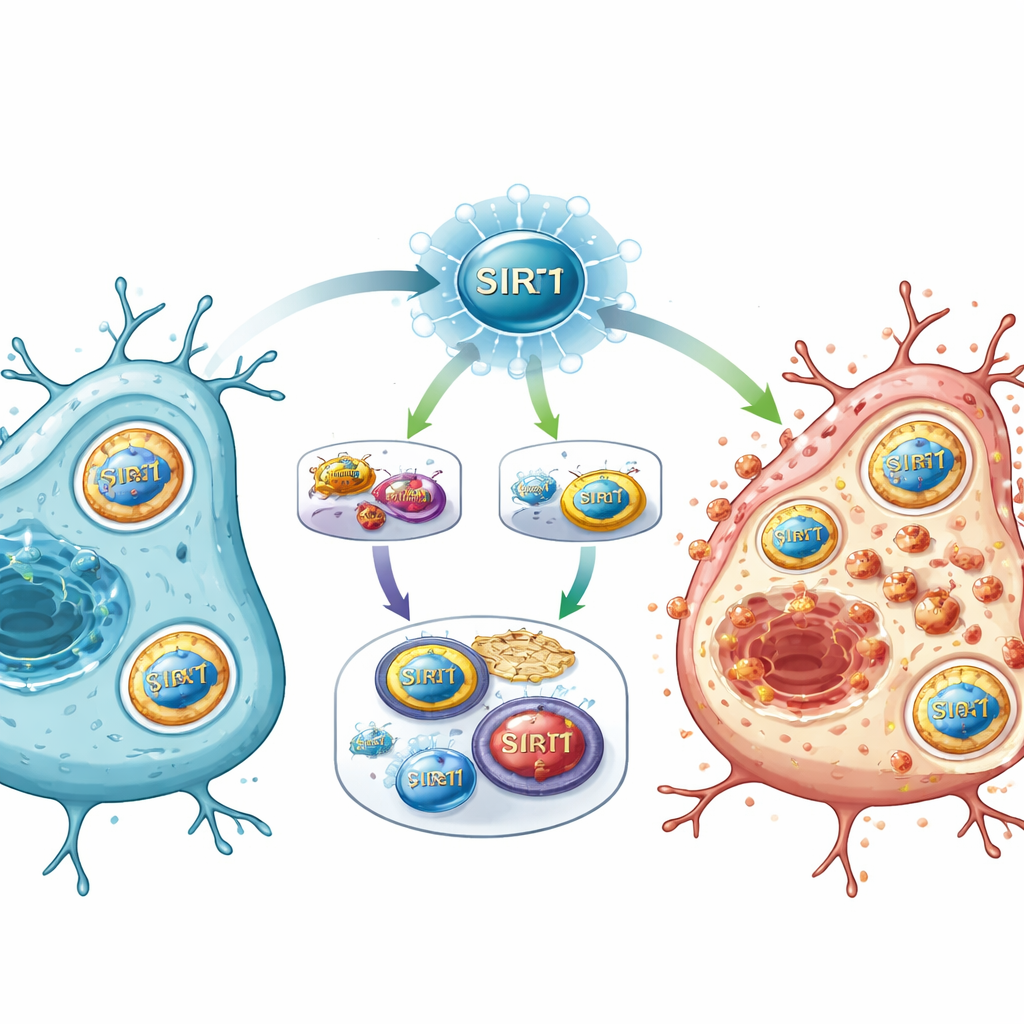
Un nouvel interrupteur maître des voies de dommage
En reconstituant les pièces du puzzle, l'étude propose que SIRT1 agit comme un interrupteur maître après l'AVC, situé au carrefour de la détection énergétique, de la régulation génique et du métabolisme du fer. Lorsqu'il est fortement activé, SIRT1 réprime la voie E2F1/Notch-1/YAP, qui aide normalement à maintenir l'expression de certains gènes de survie et des systèmes antioxydants. Simultanément, SIRT1 stimule FOXO1 et un programme d'autophagie qui recrute NCOA4 pour démanteler les protéines de stockage du fer. La surcharge en fer qui en résulte, combinée à l'affaiblissement des défenses chimiques, entraîne des dégâts lipidiques des membranes cellulaires et pousse les cellules vers la ferroptose.
Ce que cela signifie pour les soins futurs de l'AVC
Pour un non-spécialiste, le message clé est que toutes les morts cellulaires après un AVC ne se valent pas, et que certaines peuvent être évitées. Ce travail suggère que, dans le cerveau stressé, SIRT1 peut faire basculer l'équilibre vers une voie de mort dépendante du fer plutôt que vers la protection. Cibler SIRT1 lui-même, ou les étapes en aval impliquant Notch-1, YAP, FOXO1, l'autophagie et NCOA4, pourrait offrir de nouvelles façons de limiter les lésions cérébrales au-delà de la simple réouverture des vaisseaux obstrués. Bien que des études supplémentaires sur des tissus humains soient nécessaires, la voie décrite ici fournit une feuille de route pour concevoir des médicaments visant à éteindre ce feu dépendant du fer à l'intérieur des cellules cérébrales et potentiellement améliorer la récupération après un infarctus cérébral.
Citation: Wang, G., Ma, S., Qi, L. et al. SIRT1 induces FOXO1/autophagy/NCOA4-induced ferroptosis and accelerates the progression of cerebral infarction by inhibiting the E2F1/NOTCH-1/YAP signaling pathway. Sci Rep 16, 13946 (2026). https://doi.org/10.1038/s41598-026-44355-1
Mots-clés: infarctus cérébral, ferroptose, SIRT1, autophagie, mécanismes de l'accident vasculaire