Clear Sky Science · it
SIRT1 induce la ferroptosi mediata da FOXO1/autofagia/NCOA4 e accelera la progressione dell’infarto cerebrale inibendo la via di segnalazione E2F1/NOTCH-1/YAP
Perché questo studio sull’ictus è importante
L’infarto cerebrale, una forma comune di ictus, resta una delle principali cause di morte e disabilità nel mondo. I medici possono talvolta riaprire i vasi ostruiti, ma molti pazienti subiscono comunque danni cerebrali permanenti. Questo studio analizza i meccanismi cellulari per scoprire come una proteina di controllo chiave, SIRT1, possa effettivamente aggravare il danno post‑ictus spingendo le cellule verso una forma distruttiva di morte dipendente dal ferro. Comprendere questa via nascosta potrebbe aprire la strada a nuovi trattamenti che proteggano il cervello quando ogni minuto conta.
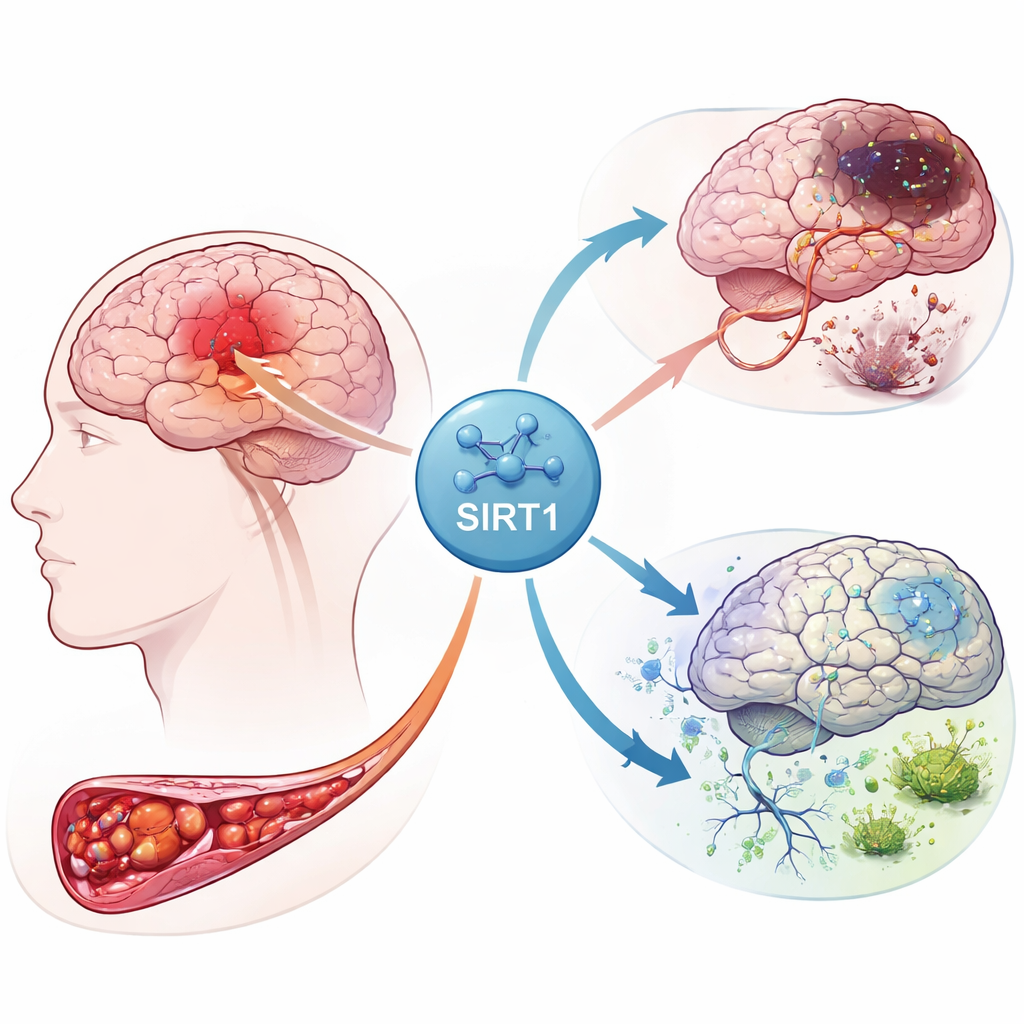
La reazione a catena all’interno di un cervello danneggiato
Quando l’apporto di sangue a una parte del cervello viene interrotto, i neuroni sono privati di ossigeno ed energia. Questa crisi scatena una tempesta di segnali in molti tipi cellulari, comprese le cellule di supporto chiamate astrociti. Gli autori si sono concentrati su una proteina chiamata SIRT1, un regolatore maestro che risponde allo stress energetico. Utilizzando dati di espressione genica provenienti da pazienti colpiti da ictus, hanno osservato che SIRT1 è strettamente correlata a geni coinvolti nella gestione del ferro, nella sopravvivenza cellulare e in una forma di morte cellulare recentemente riconosciuta chiamata ferroptosi. La loro analisi ha inoltre evidenziato connessioni tra SIRT1 e diverse vie di segnalazione già note per controllare la crescita cellulare e i processi di autolimpieza.
Dai big data al cervello animale e alle colture cellulari
Per verificare come questi indizi si manifestano nei tessuti viventi, il gruppo ha creato un modello di infarto cerebrale nel ratto bloccando brevemente una grande arteria cerebrale e poi ripristinando il flusso sanguigno. Alcuni ratti hanno ricevuto un farmaco che attiva SIRT1, mentre altri sono stati trattati con un virus che potenziava un altro componente della via, Notch‑1. Le sezioni cerebrali hanno mostrato che l’attività di SIRT1 aumentava nettamente dopo l’ictus e che l’attivazione di SIRT1 ampliava l’area di tessuto morto e aggrava il danno neuronale. Al contrario, l’aumento dei livelli di Notch‑1 riduceva il danno. I test proteici hanno confermato questo schema: quando SIRT1 era elevato, i fattori protettivi associati a Notch‑1 e alla proteina partner YAP diminuivano, mentre la lesione ischemica si espandeva.
Come ferro e autofagia guidano la morte cellulare
I ricercatori si sono poi spostati su astrociti coltivati esposti a basso ossigeno, scarse risorse nutritive e ferro aggiunto per imitare le condizioni da ictus. Manipolando con attenzione inibitori o attivatori di SIRT1, E2F1, Notch‑1, YAP e della ferroptosi, hanno mappato una sequenza dettagliata di eventi. Un alto livello di SIRT1 riduceva E2F1 nucleare, Notch‑1 e YAP, ma aumentava un altro regolatore chiamato FOXO1, insieme a proteine che segnano un incremento dell’autodigestione (autofagia) e a un adattatore di carico denominato NCOA4. Questa combinazione favoriva un tipo specifico di autofagia che smonta i complessi di deposito del ferro, inondando le cellule di ferro libero. In parallelo, i marcatori delle difese antiossidanti cambiavano e la morte cellulare aumentava, corrispondendo alla firma biochimica della ferroptosi. Bloccare la ferroptosi o ripristinare parti dell’asse Notch‑1/YAP attenuava questi cambiamenti dannosi.
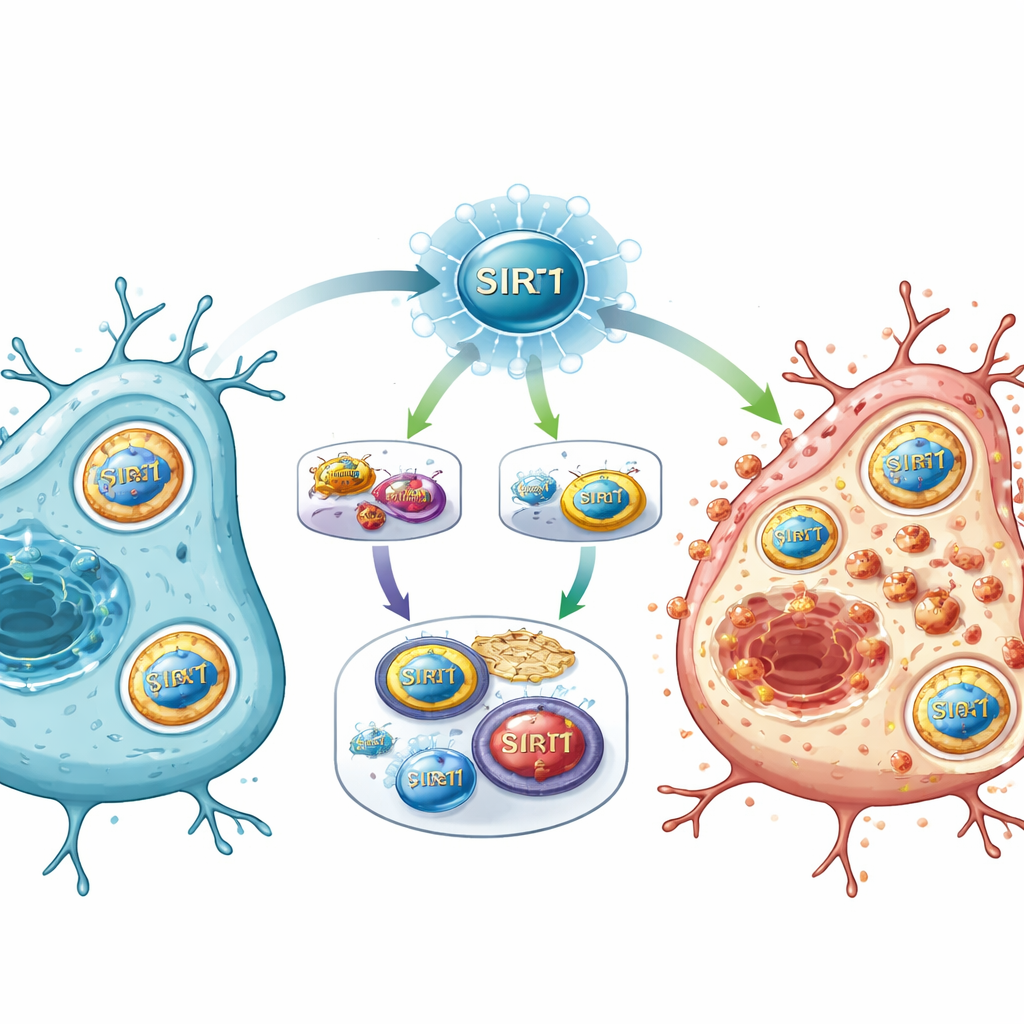
Un nuovo interruttore maestro per le vie del danno
Combinando i risultati, lo studio propone che SIRT1 agisca come un interruttore maestro dopo l’ictus, posizionandosi all’incrocio tra la rilevazione energetica, la regolazione genica e il metabolismo del ferro. Quando SIRT1 è fortemente attivato, sopprime la via E2F1/Notch‑1/YAP, che normalmente contribuisce a mantenere attivi alcuni geni di sopravvivenza e i sistemi antiossidanti. Allo stesso tempo, SIRT1 potenzia FOXO1 e un programma di autofagia che recluta NCOA4 per smantellare le proteine di stoccaggio del ferro. Il conseguente sovraccarico di ferro, insieme all’indebolimento delle difese chimiche, promuove il danneggiamento lipidico nelle membrane cellulari e spinge le cellule verso la ferroptosi.
Cosa significa per la cura futura dell’ictus
Per il lettore non specialista, il messaggio chiave è che non tutte le morti cellulari dopo un ictus sono uguali e parte di esse potrebbe essere prevenibile. Questo lavoro suggerisce che, nel cervello sotto stress, SIRT1 può sbilanciare il sistema verso una via di morte dipendente dal ferro anziché verso la protezione. Mirare a SIRT1 stesso, o ai passaggi a valle che coinvolgono Notch‑1, YAP, FOXO1, l’autofagia e NCOA4, potrebbe offrire nuovi modi per limitare il danno cerebrale oltre la semplice riapertura dei vasi ostruiti. Pur richiedendo ulteriori studi su tessuti umani, la via delineata qui fornisce una mappa per progettare farmaci che raffreddino questo fuoco guidato dal ferro nelle cellule cerebrali e migliorino potenzialmente il recupero dopo un infarto cerebrale.
Citazione: Wang, G., Ma, S., Qi, L. et al. SIRT1 induces FOXO1/autophagy/NCOA4-induced ferroptosis and accelerates the progression of cerebral infarction by inhibiting the E2F1/NOTCH-1/YAP signaling pathway. Sci Rep 16, 13946 (2026). https://doi.org/10.1038/s41598-026-44355-1
Parole chiave: infarto cerebrale, ferroptosi, SIRT1, autofagia, meccanismi dell’ictus