Clear Sky Science · pl
SIRT1 indukuje FOXO1/autofagię/NCOA4‑zależną ferroptozę i przyspiesza progresję zawału mózgu poprzez hamowanie szlaku sygnałowego E2F1/NOTCH‑1/YAP
Dlaczego to badanie nad udarem ma znaczenie
Zawał mózgu, powszechna postać udaru, pozostaje jedną z głównych przyczyn zgonów i niepełnosprawności na świecie. Lekarzom czasem udaje się przywrócić drożność zatkanych naczyń, ale wielu chorych i tak pozostaje z trwałym uszkodzeniem mózgu. Badanie to zagląda do wnętrza komórek mózgowych, aby ujawnić, jak kluczowe białko regulatorowe SIRT1 może w rzeczywistości pogarszać uraz po udarze, popychając komórki w stronę destrukcyjnej formy śmierci zależnej od żelaza. Zrozumienie tej ukrytej ścieżki może otworzyć drogę do nowych terapii chroniących mózg, gdy każda minuta ma znaczenie.
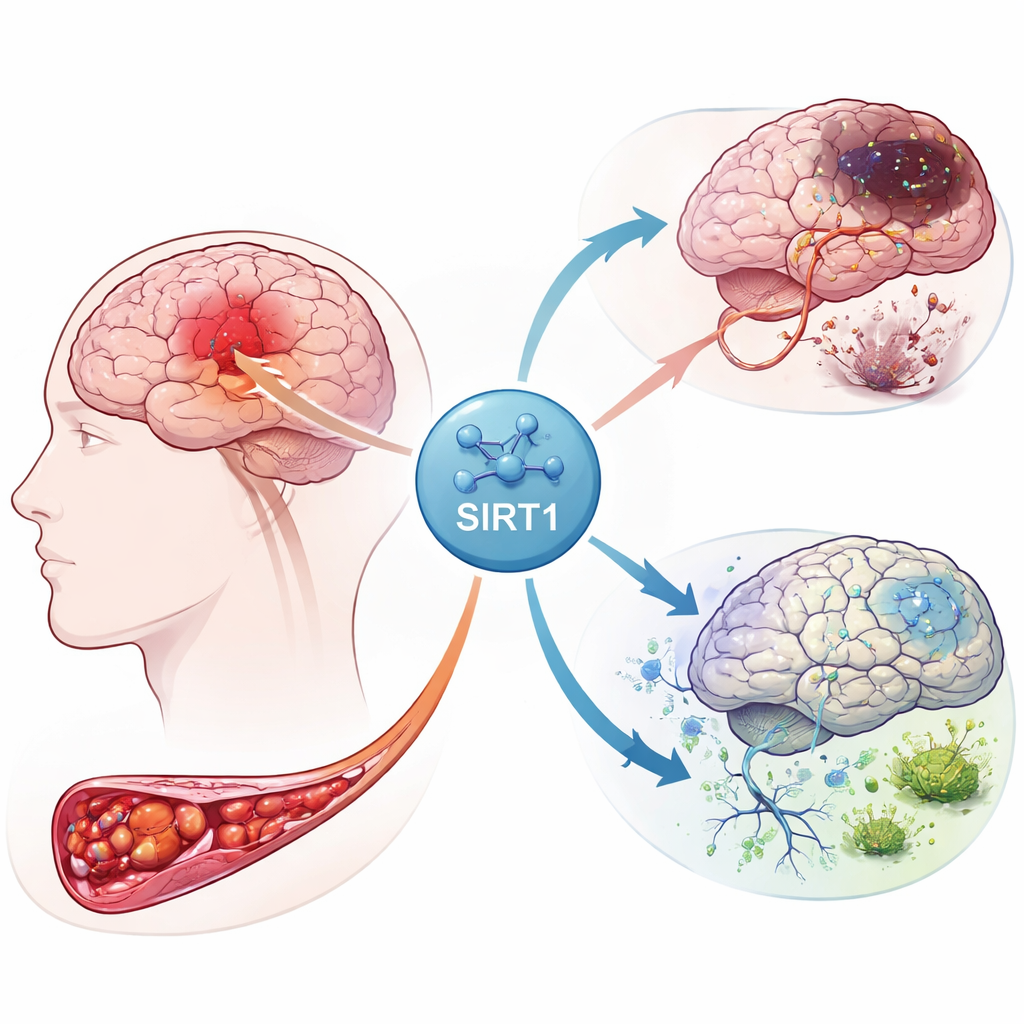
Reakcja łańcuchowa we wnętrzu uszkodzonego mózgu
Gdy dopływ krwi do części mózgu zostaje przerwany, neurony pozbawione są tlenu i energii. Ten kryzys wyzwala burzę sygnałów w wielu typach komórek, w tym w komórkach podporowych zwanych astrocytami. Autorzy skoncentrowali się na białku SIRT1, głównym regulatorze reagującym na stres energetyczny. Analizując dane ekspresji genów od pacjentów po udarze, stwierdzili, że SIRT1 jest silnie powiązany z genami uczestniczącymi w gospodarce żelaza, przetrwaniu komórek oraz z nowo rozpoznaną formą śmierci komórkowej – ferroptozą. Ich analiza podkreśliła też połączenia między SIRT1 a kilkoma szlakami sygnałowymi już znanymi z kontroli wzrostu komórek i procesów samoczyszczenia.
Od big data do mózgów zwierząt i hodowli komórek
Aby sprawdzić, jak te wskazówki sprawdzają się w żywej tkance, zespół stworzył model zawału mózgu u szczurów przez krótkotrwałe zablokowanie dużej tętnicy mózgowej, a następnie przywrócenie przepływu krwi. Niektóre zwierzęta otrzymały lek aktywujący SIRT1, inne zaś wirusa zwiększającego poziom komponentu szlaku, Notch‑1. Preparaty mózgu wykazały, że aktywność SIRT1 gwałtownie wzrosła po udarze, a jej zwiększenie powiększało obszar martwicy i nasilenie uszkodzeń neuronów. W przeciwieństwie do tego podwyższenie poziomu Notch‑1 zmniejszało rozległość uszkodzeń. Badania białek potwierdziły ten wzór: przy wysokim poziomie SIRT1 spadały czynniki ochronne związane z Notch‑1 i białkiem współpracującym YAP, a uraz udarowy się rozszerzał.
Jak żelazo i „samozjadanie” napędzają śmierć komórki
Następnie badacze przeszli do hodowli astrocytów wystawionych na niskie stężenie tlenu, niedobór składników odżywczych i dodane żelazo, aby naśladować warunki po udarze. Poprzez ostrożne stosowanie inhibitorów lub aktywatorów SIRT1, E2F1, Notch‑1, YAP i ferroptozy, zmapowali szczegółowy łańcuch zdarzeń. Wysoki poziom SIRT1 obniżał jądrowy E2F1, Notch‑1 i YAP, jednocześnie zwiększając poziom innego regulatora, FOXO1, oraz białek wskazujących na wzmożoną autodiagnagę (autofagię) i adaptor ładunku NCOA4. To połączenie sprzyjało specyficznej formie autofagii, która rozbija kompleksy magazynujące żelazo, zalewając komórki wolnym żelazem. W miarę jak to następowało, zmieniały się markery obrony antyoksydacyjnej, a śmierć komórek rosła, odpowiadając biochemicznemu podpisowi ferroptozy. Zablokowanie ferroptozy lub przywrócenie części osi Notch‑1/YAP ograniczało te szkodliwe zmiany.
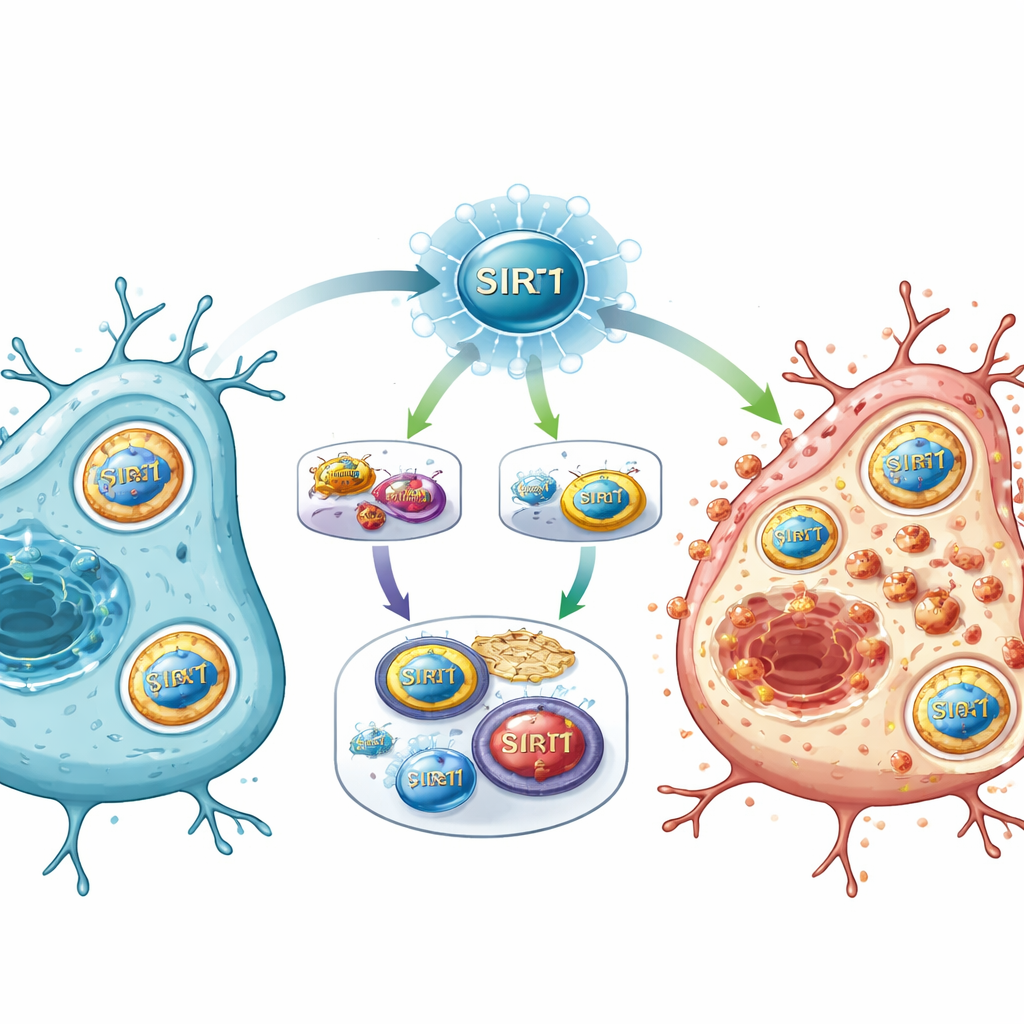
Nowy główny przełącznik ścieżek urazowych
Składając elementy w całość, badanie proponuje, że SIRT1 działa jako główny przełącznik po udarze, znajdując się na skrzyżowaniu wyczuwania energii, regulacji genów i metabolizmu żelaza. Gdy SIRT1 jest silnie aktywowany, tłumi szlak E2F1/Notch‑1/YAP, który normalnie pomaga utrzymać działanie genów przetrwania i systemów antyoksydacyjnych. Jednocześnie SIRT1 wzmacnia FOXO1 i program autofagii rekrutujący NCOA4 do rozkładu białek magazynujących żelazo. W efekcie dochodzi do przeciążenia żelazem, a osłabione mechanizmy obronne sprzyjają uszkodzeniom lipidów w błonach komórkowych i popychają komórki w stronę ferroptozy.
Co to oznacza dla przyszłej opieki po udarze
Dla czytelnika niebędącego specjalistą kluczowa wiadomość jest taka, że nie każda śmierć komórek po udarze jest taka sama i część z niej może być możliwa do powstrzymania. Praca ta sugeruje, że w zestresowanym mózgu SIRT1 może przechylić równowagę w kierunku destrukcyjnej, zależnej od żelaza ścieżki śmierci zamiast ochrony. Celowanie w sam SIRT1 lub kroki zstępujące obejmujące Notch‑1, YAP, FOXO1, autofagię i NCOA4 mogłoby dać nowe możliwości ograniczenia uszkodzeń mózgu poza samym przywracaniem przepływu krwi. Choć potrzebne są dalsze badania na tkankach ludzkich, tu przedstawiony szlak oferuje mapę drogową do projektowania leków, które stłumią ten żelazny pożar wewnątrz komórek mózgowych i potencjalnie poprawią rekonwalescencję po zawale mózgu.
Cytowanie: Wang, G., Ma, S., Qi, L. et al. SIRT1 induces FOXO1/autophagy/NCOA4-induced ferroptosis and accelerates the progression of cerebral infarction by inhibiting the E2F1/NOTCH-1/YAP signaling pathway. Sci Rep 16, 13946 (2026). https://doi.org/10.1038/s41598-026-44355-1
Słowa kluczowe: zawał mózgu, ferroptoza, SIRT1, autofagia, mechanizmy udaru