Clear Sky Science · es
SIRT1 induce ferroptosis mediada por FOXO1/autofagia/NCOA4 y acelera la progresión del infarto cerebral al inhibir la vía de señalización E2F1/NOTCH-1/YAP
Por qué importa este estudio sobre el ictus
El infarto cerebral, una forma común de ictus, sigue siendo una de las principales causas de muerte y discapacidad en todo el mundo. A veces los médicos pueden reabrir vasos sanguíneos obstruidos, pero muchos pacientes quedan con daño cerebral permanente. Este estudio examina el interior de las células cerebrales para descubrir cómo una proteína reguladora clave, SIRT1, puede en realidad empeorar la lesión tras un ictus al impulsar a las células hacia una forma destructiva de muerte dependiente del hierro. Comprender esta vía oculta podría abrir la puerta a nuevos tratamientos que protejan el cerebro cuando cada minuto cuenta.
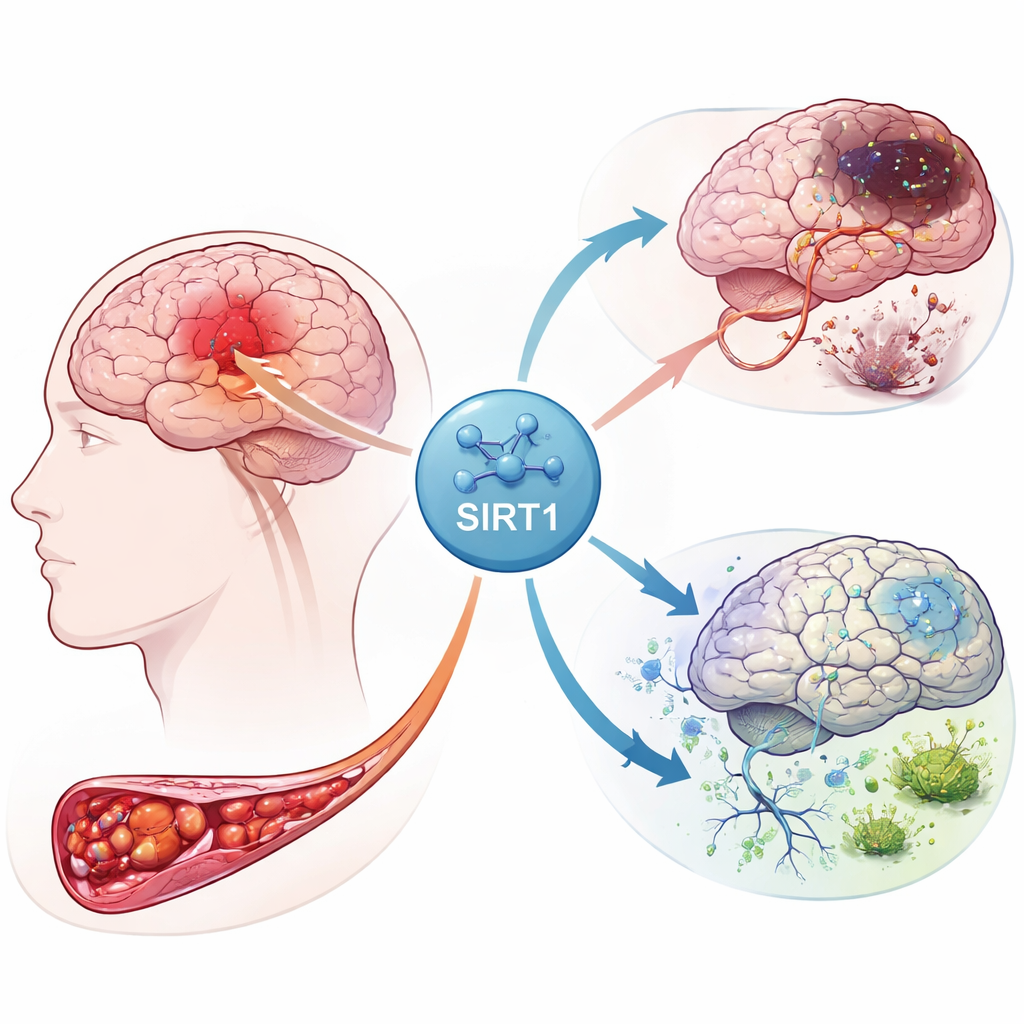
La reacción en cadena dentro de un cerebro dañado
Cuando el flujo sanguíneo a una parte del cerebro se corta, las neuronas se quedan sin oxígeno ni energía. Esta crisis desencadena una tormenta de señales dentro de muchos tipos celulares, incluidas las células gliales llamadas astrocitos. Los autores se centraron en una proteína llamada SIRT1, un regulador maestro que responde al estrés energético. Usando datos de expresión génica de pacientes con ictus, encontraron que SIRT1 se asociaba estrechamente con genes implicados en el manejo del hierro, la supervivencia celular y una forma de muerte celular recientemente reconocida llamada ferroptosis. Su análisis también resaltó conexiones entre SIRT1 y varias rutas de señalización ya conocidas por controlar el crecimiento celular y los procesos de limpieza interna.
De los macrodatos al cerebro animal y los cultivos celulares
Para ver cómo se manifiestan estas pistas en tejido vivo, el equipo creó un modelo de ratas con infarto cerebral bloqueando brevemente una arteria cerebral principal y luego restaurando el flujo sanguíneo. Algunas ratas recibieron un fármaco que activa SIRT1, mientras que a otras se les administró un virus que aumentaba otro componente de la vía, Notch-1. Los cortes cerebrales mostraron que la actividad de SIRT1 aumentó notablemente después del ictus y que la activación de SIRT1 amplificó el área de tejido muerto y el daño neuronal. En contraste, elevar los niveles de Notch-1 redujo el daño. Las pruebas de proteínas confirmaron este patrón: cuando SIRT1 era alto, los factores protectores asociados con Notch-1 y una proteína compañera, YAP, disminuían, mientras la lesión por ictus se expandía.
Cómo el hierro y la autodegradación impulsan la muerte celular
Los investigadores continuaron con astrocitos cultivados expuestos a bajo oxígeno, escasos nutrientes y aporte adicional de hierro para imitar las condiciones del ictus. Mediante la administración controlada de inhibidores o activadores de SIRT1, E2F1, Notch-1, YAP y de la ferroptosis, trazaron una cadena de eventos detallada. SIRT1 elevado reprimió E2F1 nuclear, Notch-1 y YAP, pero aumentó otro regulador llamado FOXO1, junto con proteínas que indican una mayor autodegradación (autofagia) y un adaptador de carga denominado NCOA4. Esta combinación favoreció un tipo específico de autofagia que desmonta los complejos de almacenamiento de hierro, inundando las células con hierro libre. A medida que esto ocurría, los marcadores de defensa antioxidante se desplazaron y la muerte celular se disparó, coincidiendo con la firma bioquímica de la ferroptosis. Bloquear la ferroptosis o restaurar partes del eje Notch-1/YAP redujo estos cambios dañinos.
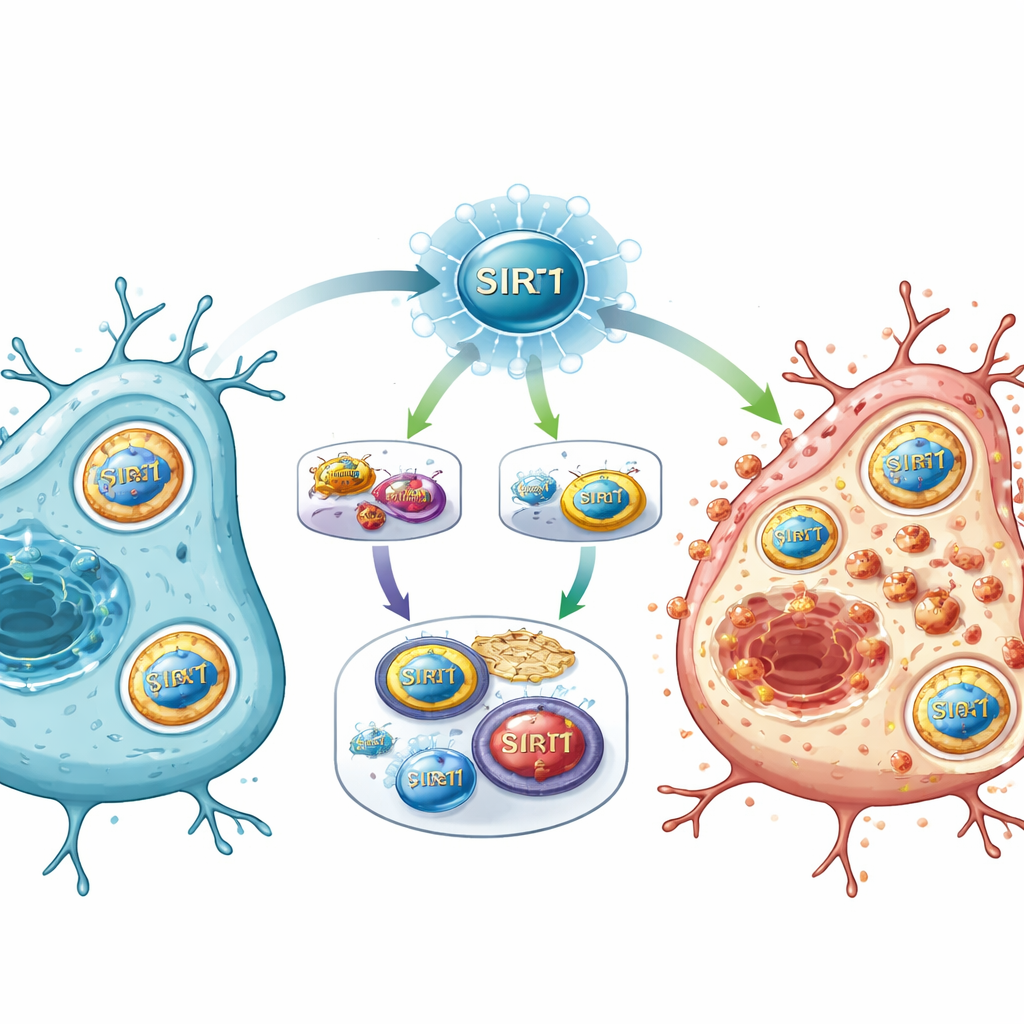
Un nuevo interruptor maestro para las vías de lesión
Al reunir las piezas, el estudio propone que SIRT1 actúa como un interruptor maestro tras el ictus, situado en la encrucijada entre la detección energética, la regulación génica y el metabolismo del hierro. Cuando SIRT1 se activa con fuerza, reprime la vía E2F1/Notch-1/YAP, que normalmente ayuda a mantener en funcionamiento ciertos genes de supervivencia y los sistemas antioxidantes. Al mismo tiempo, SIRT1 potencia FOXO1 y un programa de autofagia que recluta a NCOA4 para desmantelar las proteínas de almacenamiento del hierro. La sobrecarga resultante de hierro, combinada con defensas químicas debilitadas, provoca daño lipídico en las membranas celulares y empuja a las células hacia la ferroptosis.
Qué significa esto para la atención futura del ictus
Para un público no especialista, el mensaje clave es que no toda la muerte celular tras un ictus es igual, y parte de ella podría ser prevenible. Este trabajo sugiere que, en el cerebro estresado, SIRT1 puede inclinar la balanza hacia una vía de muerte dependiente del hierro en lugar de la protección. Dirigirse a SIRT1 en sí, o a los pasos aguas abajo que implican Notch-1, YAP, FOXO1, la autofagia y NCOA4, podría ofrecer nuevas maneras de limitar el daño cerebral más allá de la mera reapertura de vasos bloqueados. Aunque se necesitan más estudios en tejido humano, la vía descrita aquí proporciona una hoja de ruta para diseñar fármacos que atenúen este fuego impulsado por el hierro dentro de las células cerebrales y potencialmente mejoren la recuperación tras el infarto cerebral.
Cita: Wang, G., Ma, S., Qi, L. et al. SIRT1 induces FOXO1/autophagy/NCOA4-induced ferroptosis and accelerates the progression of cerebral infarction by inhibiting the E2F1/NOTCH-1/YAP signaling pathway. Sci Rep 16, 13946 (2026). https://doi.org/10.1038/s41598-026-44355-1
Palabras clave: infarto cerebral, ferroptosis, SIRT1, autofagia, mecanismos del ictus