Clear Sky Science · zh
通过张量分解在全基因组 CRISPR-Cas9 敲除筛选中计算基因和细胞系效率
寻找我们 DNA 工具箱中的关键部分
CRISPR 基因编辑已成为现代生物学中最强大的工具之一,允许科学家同时关闭成千上万个基因,以观察哪些基因对细胞存活真正重要。但将这类大量实验数据转化为明确结论却出奇地困难。本文介绍了一种简单却有效的数学方法,称为张量分解,它可帮助科学家更可靠地解读这些大规模 CRISPR 实验,即便一些常见的对照样本缺失。
为什么让基因失活会如此混乱
在典型的全基因组 CRISPR 实验中,研究者使用许多短的引导分子(称为 sgRNA)在多种不同细胞系中敲除每个基因。理论上,如果丢失某个基因会导致细胞死亡或功能受损,该基因就是必需的;如果没有明显影响,则可能是非必需的。实际上,每个引导的有效性各不相同,不同实验室对结果的测量方式也略有差异。因此,科学家必须以某种方式将来自多重引导、基因和细胞系的嘈杂读数合并为一个单一评分,以判断每个基因的真实重要性。当前许多方法通过精细的统计模型来实现这一点,且常常需要特殊的对照样本作为锚点。
一种同时从多个方向观察数据的简单方法
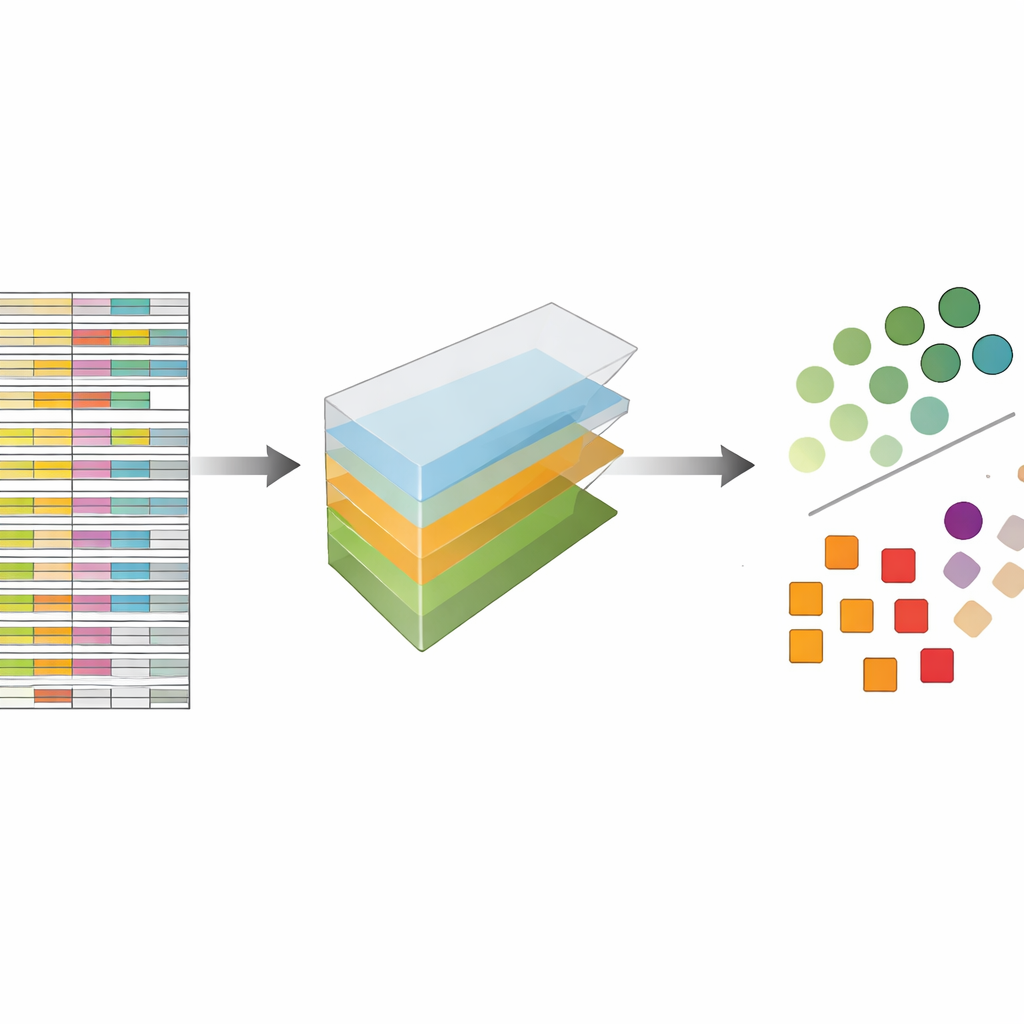
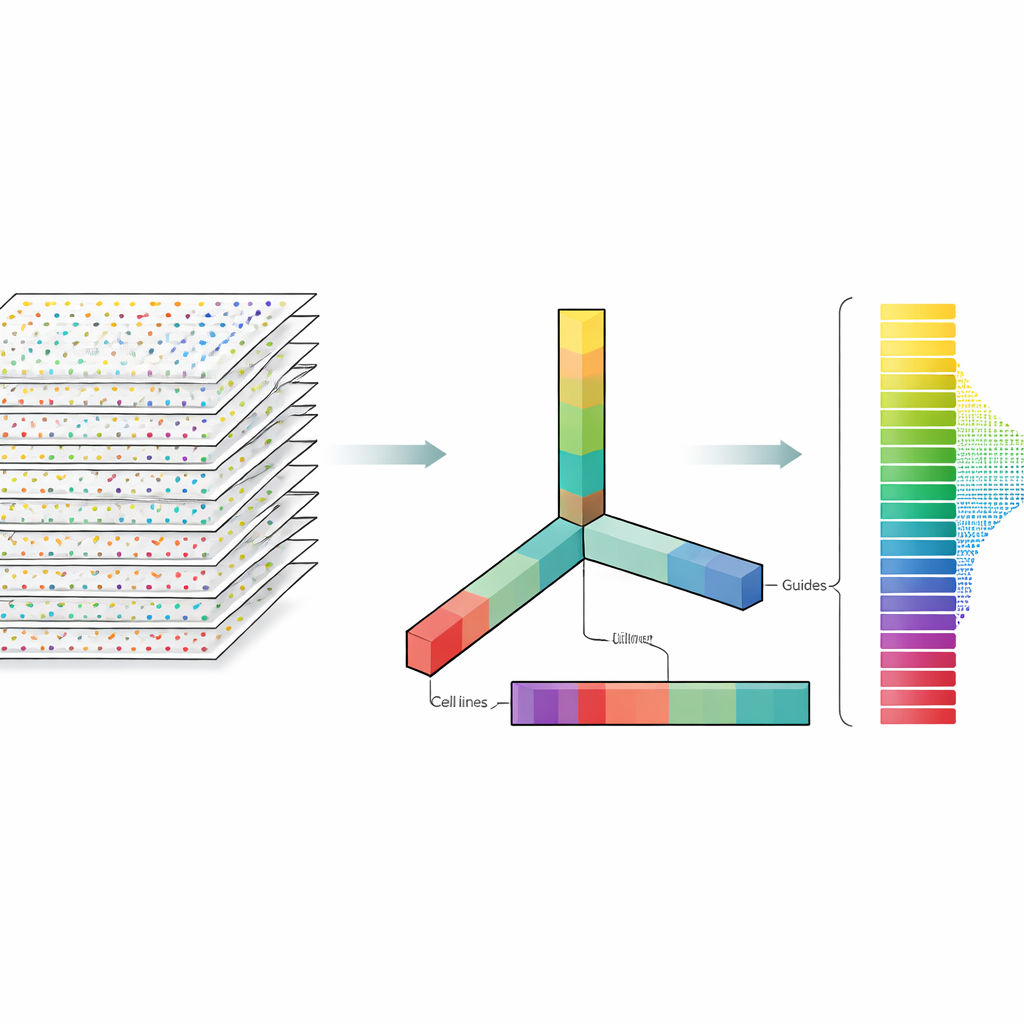
作者将 CRISPR 数据视为不是扁平的电子表格,而是可以沿多个方向切片的多维数据块:基因、引导、细胞系和实验重复。张量分解是一种线性代数技术,可将该数据块分解为一组基本模式及其权重,用以表示每种模式出现的强度。无需先验标签或训练,这些模式就能突出显示哪些基因类似已知的必需基因,哪些细胞系具有相似的反应模式。关键在于,该方法从一开始就综合考虑了每基因的多个引导和多个实验配置,而不是分别分析每个文件再在后期合并结果。
在不复杂化的情况下匹配最先进方法的表现
为检验该方法的效果,作者将其应用于五个大型且广泛使用的 CRISPR 筛选集合,这些数据集此前已用一种名为 JACKS 的领先方法以及其他先进工具进行过分析。他们提出了一个简单问题:他们的方法能在多大程度上区分已发表的必需基因列表与非必需基因?以标准准确度指标“曲线下面积”(AUC)衡量,张量分解在各数据集上的表现与 JACKS 不相上下,常常达到约 0.8 的值,这在该领域被认为是很强的表现。更引人注目的是,性能曲线的详细形状与 JACKS 十分一致,这表明这种更简单的方法捕捉到了与更复杂贝叶斯方法大部分相同的生物学信号。
在缺少对照且使用原始计数时也能工作
部分数据集缺乏许多方法依赖的常规对照样本,但张量分解仍表现良好。在有对照的数据集中,该方法自然能够挑出将对照样本与处理样本分开的模式,从而帮助识别必需基因。在无对照的数据集中,它则发现了与各细胞系中 CRISPR 效率的独立测量高度一致的模式,这些效率测量来自一个大型癌症依赖性项目。另一个实用上的意外是,该方法在使用原始计数数据时的表现与在使用对数变换后的数据时相当,而对数变换是一种常见但并非总是合理的预处理步骤。这一发现表明,CRISPR 筛选可能并不需要像通常假定的那样进行大量的数值处理。
这对未来基因编辑研究意味着什么
总体而言,该研究表明,一种相对简单的数学透镜能够与用于分析大规模 CRISPR 筛选的复杂、高度调优模型并肩。通过同时处理众多引导和多个实验,张量分解可以可靠地区分必需与非必需基因,并揭示不同细胞系中 CRISPR 效率的差异,即便没有理想的对照。对非专业读者来说,关键结论是:以更聪明的方式观察相同数据,可以使基因编辑实验更值得信赖、更易比较,从而帮助研究者更快地识别对健康与疾病最重要的基因。
引用: Taguchi, YH., Turki, T. Gene and cell line efficiency of CRISPR computed by tensor decomposition in genome-wide CRISPR-Cas9 knockout screens. Sci Rep 16, 13605 (2026). https://doi.org/10.1038/s41598-026-43209-0
关键词: CRISPR 筛选, 基因必需性, 张量分解, sgRNA 效率, 癌症细胞系