Clear Sky Science · en
Gene and cell line efficiency of CRISPR computed by tensor decomposition in genome-wide CRISPR-Cas9 knockout screens
Finding the Important Parts of Our DNA Toolkit
CRISPR gene editing has become one of the most powerful tools in modern biology, letting scientists switch off thousands of genes at once to see which ones truly matter for cell survival. But turning that flood of experimental data into clear answers is surprisingly hard. This paper introduces a simple yet effective mathematical approach, called tensor decomposition, that helps scientists read these massive CRISPR experiments more reliably, even when some of the usual control samples are missing.
Why Turning Genes Off Is So Messy
In a typical genome-wide CRISPR experiment, researchers use many short guide molecules, called sgRNAs, to knock out each gene in many different cell lines. In theory, if losing a gene kills or weakens cells, that gene is essential; if nothing much happens, it is probably non-essential. In practice, each guide varies in how well it works, and different labs measure results in slightly different ways. As a result, scientists must somehow combine the noisy readouts from many guides, genes, and cell lines into a single score that says how important each gene really is. Many current methods do this with elaborate statistical models and often require special control samples as anchors.
A Simple Way to Look at Data in Several Directions at Once
The authors treat CRISPR data not as a flat spreadsheet but as a multi-dimensional block they can slice along several directions at once: genes, guides, cell lines, and experimental repeats. Tensor decomposition is a linear algebra technique that breaks this block into a set of basic patterns plus weights that say how strongly each pattern appears. Without needing prior labels or training, these patterns can highlight which genes behave like known essential genes and which cell lines share similar response patterns. Crucially, the method integrates both multiple guides per gene and multiple experimental profiles from the very beginning, instead of analyzing each file separately and merging results later.
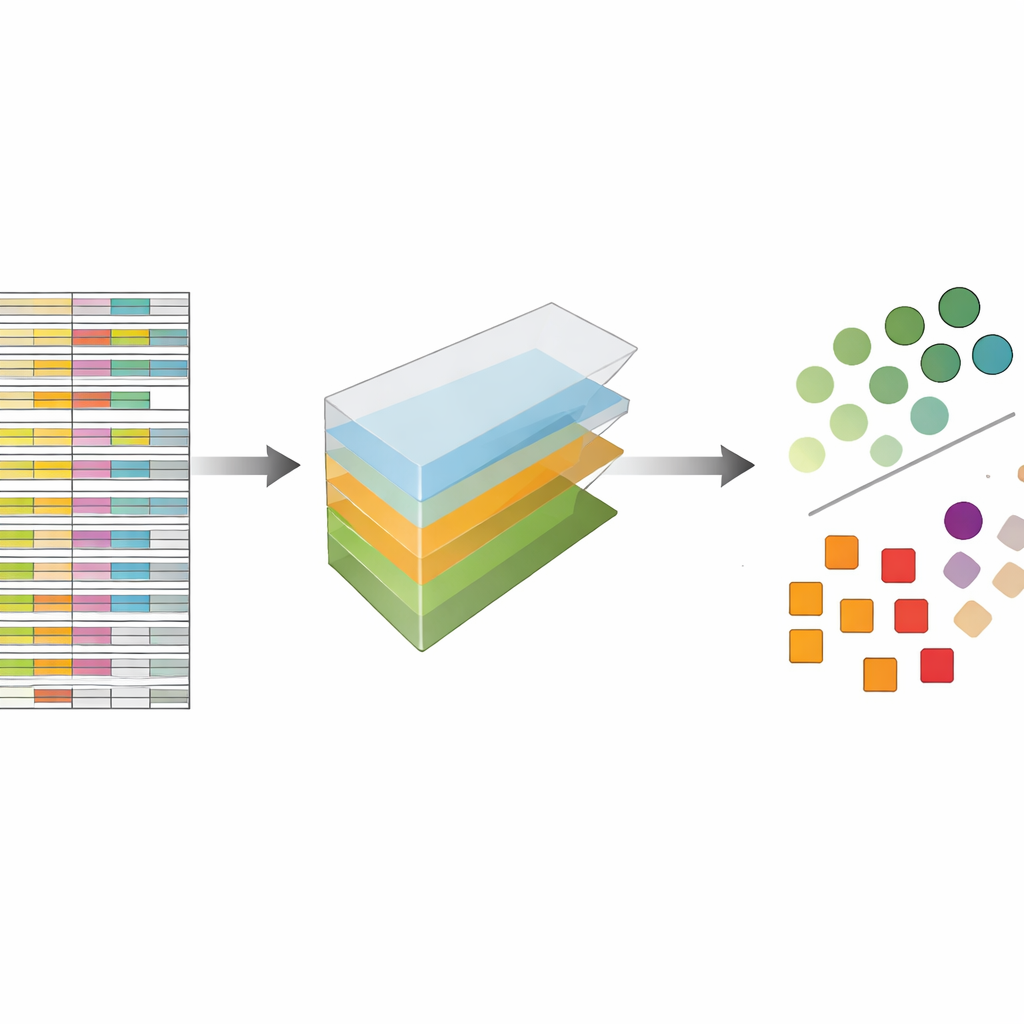
Matching the Best Modern Methods Without Extra Bells and Whistles
To test how well this approach works, the authors applied it to five large, widely used CRISPR screening collections that had previously been analyzed with a leading method called JACKS, as well as other state-of-the-art tools. They asked a simple question: how well can their method distinguish a published list of essential genes from non-essential ones? Measured by a standard accuracy score known as the area under the curve, tensor decomposition performed on par with JACKS across datasets, often reaching values around 0.8, which is considered strong in this context. Even more striking, the detailed shapes of the performance curves closely matched JACKS, suggesting that the simpler method is capturing much of the same biological signal as the more complex Bayesian approach.
Working When Controls Are Missing and Numbers Stay Raw
Some of the datasets lacked the usual control samples that many methods rely on, yet tensor decomposition still worked well. In datasets with controls, the method naturally picked out patterns that separated control samples from treated ones, which helped it find essential genes. In datasets without controls, it instead discovered patterns that closely tracked independent measures of how efficiently CRISPR works in each cell line, drawn from a large cancer dependency project. Another practical surprise was that the method did just as well when using the raw count data as when using logarithmic transformations, a common but not always justified preprocessing step. This finding raises the possibility that CRISPR screens may not need as much numerical massaging as is often assumed.
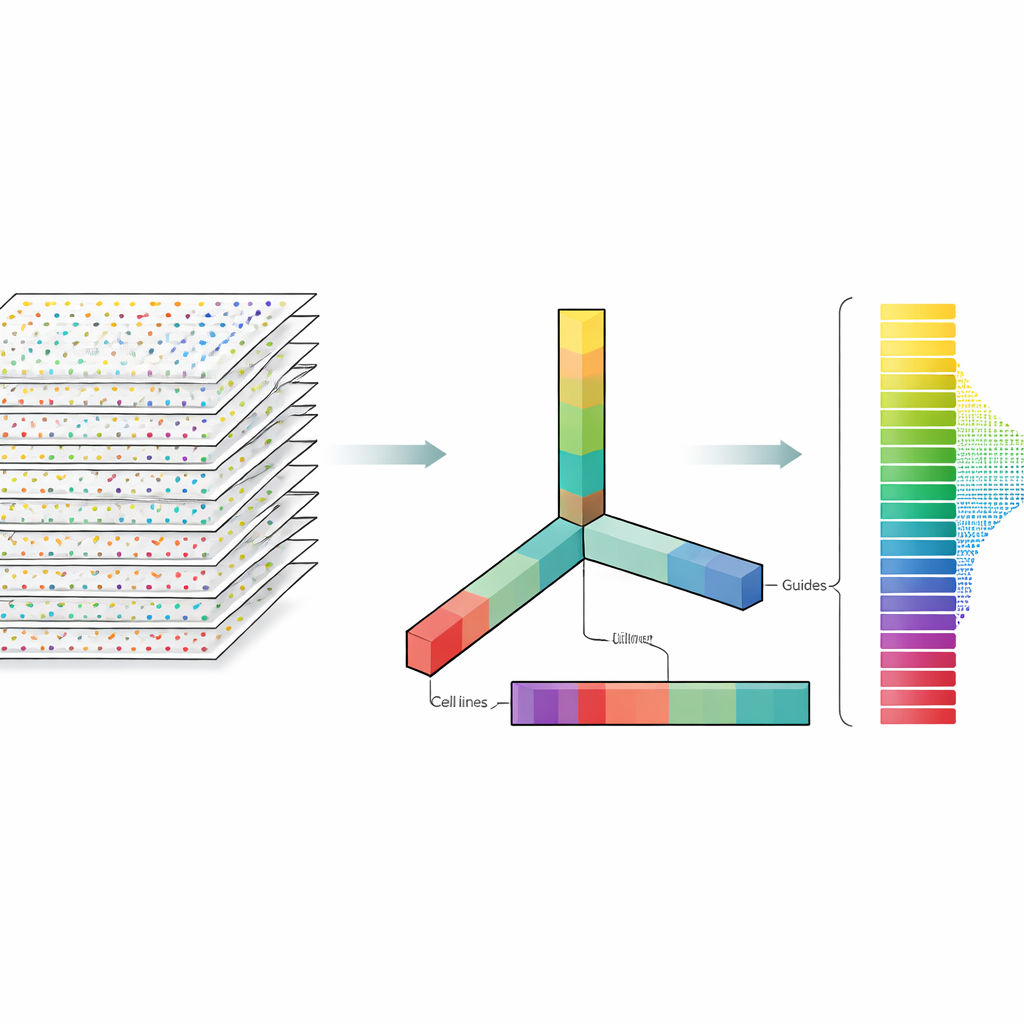
What This Means for Future Gene-Editing Studies
Overall, the study shows that a relatively simple mathematical lens can stand shoulder to shoulder with sophisticated, highly tuned models for analyzing large-scale CRISPR screens. By jointly handling many guides and many experiments at once, tensor decomposition can reliably separate essential from non-essential genes and reveal differences in how well CRISPR works across cell lines, even without ideal controls. For non-specialists, the key message is that smarter ways of looking at the same data can make gene-editing experiments more trustworthy and easier to compare, helping researchers more quickly identify the genes that matter most for health and disease.
Citation: Taguchi, YH., Turki, T. Gene and cell line efficiency of CRISPR computed by tensor decomposition in genome-wide CRISPR-Cas9 knockout screens. Sci Rep 16, 13605 (2026). https://doi.org/10.1038/s41598-026-43209-0
Keywords: CRISPR screens, gene essentiality, tensor decomposition, sgRNA efficiency, cancer cell lines